|
||||||||||
Date: June 20, 2024
by Chaya Venkat
Related Article:

Yet another new molecule has appeared on the horizon. As they say, hope springs eternal, especially in cancer patients and their families. This molecule (and its kissing cousins) has been the subject of much pre-clinical scrutiny, and I have seen recent announcements of several Phase I / II clinical trials in a variety of solid cancers as well as a couple of blood cancers. It is too soon to tell how it performed in these early human clinical trials. I have seen very sparse data on this drug with reference to CLL, not even mouse model studies yet. However, I am pretty sure we will be seeing clinical trials of this drug in live CLL patients before long at one of our more cutting edge research centers. I am sure you will agree it is time to learn a little bit about this drug, before you sign your name on the dotted line at the bottom of the consent form.
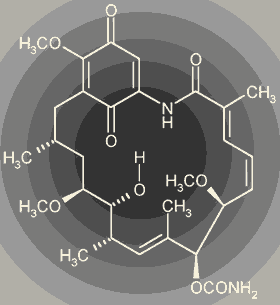 |
Geldanamycin is a natural product produced by Streptomyces hygroscopicus. It belongs to a family of antibiotics that were later shown to have potent anti-cancer activity. In the months to come I suspect you will be seeing the name of its more recent analog labeled "17-AAG" (trust me, you really do not want to know what the full name is, it is longer than most Indian names that I have come across). 17-AAG is thought to have equal or better potency than geldanamycin as a cancer cell killer, without some of the toxicity associated with the parent molecule.
Proteins are involved in just about every aspect of the functioning of our bodies. The instructions for making the right proteins are encoded in each and every one of our cells. The various genes in the chromosomes make all the different proteins we need to carry on daily functioning of our bodies. As you should know by now, CLL cells can have a variety of defects in the chromosomes with different genes deleted, mutated or just plain covered over with methyl gunk and therefore unable to function (FISHing for Answers; ATM and P53: Gatekeepers of the Genome; Epigenetics).
However, producing the right proteins is only the first step; an equally important step is making sure they are folded just right. Proteins are very large and complex molecules, with a zillion arms and legs, and they have to be folded into exactly the right three-dimensional shapes before they can do their jobs. I was staying at a large hotel the other day, and sat watching the housekeeping staff make my bed the next morning. The cart had a huge stack of neatly ironed and folded bed linens. The maid picked up one of the sheets, and as it happened the sheet was not ironed right and miss-folded. Without a second look at it she pitched the offending bed sheet into the used linen bin, and grabbed a new one. The same thing happens to miss-folded proteins. There are garbage pick-up molecules called proteosomes trolling around, whose job is to dispose / destroy improperly folded proteins.
How do proteins get folded? They cannot do it all by themselves, they need help. The “chaperone” molecules that supervise folding of proteins are called “Heat Shock Proteins”, Hsp for short. There is one particular heat shock protein, called Hsp90, that is very important. Think of it as the team leader, it controls the functioning of a whole series of lesser heat shock proteins. All in all, Hsp90 is involved in the proper folding of a large list of “client” proteins. Since cancer cells typically have messed up genes, and therefore produce messed up proteins, they compensate for it by having an extra helping of Hsp90. The result is that even improperly made proteins get folded up sufficiently well to fool the garbage disposal squad. Hsp90 supervises the proper folding of a variety of mutated and over-expressed proteins that promote growth and survival of cancer cells. Among Hsp90’s “client proteins” are mutated p53 (important in CLL and most other cancers), Raf-1, Akt, Her2/Neu (breast cancer), Bcr-Abl (important in acute lymphoblastic leukemia, ALL), etc. It has been demonstrated that majority of cancer cells express copious amounts of Hsp90 as a way of compensating for the malformed proteins and allow the cancer cells to continue living.
What would happen to a cancer cell if we were to block the activity of the Hsp90, the “chaperone” that makes sure its important client proteins are folded right? For starters, the messed up proteins produced by the cancer cell would not fold right, and would not function right. These gobs of useless proteins are quickly disposed of by proteosomes, the bodies version of garbage disposals. Deprived of the function of these essential proteins, the cell dies quickly. Blocking the function of Hsp90 is at the heart of the potential usefulness of 17-AAG in cancer therapy. Since cancer cells have a higher dependence on Hsp90 than normal cells, it makes this heat shock protein an attractive target. It turns out that 17-AAG has the right shape to bind to an important “pocket” in the structure of Hsp90, taking it out of play and rendering it unable to assist in the folding of proteins. We hope this will interfere with the functioning of cancer cells on a variety of different and important fronts, a multi-pronged approach that may be more effective than targeting a single point of vulnerability.
17-AAG cannot tell the difference between Hsp90 expressed by a healthy cell and that expressed by a cancer cell. But if our theory is right, since cancer cells are so much more dependent upon the functioning of Hsp90, blocking it may hurt them a lot more than it would hurt normal cells. The whole trade-off between efficacy and toxicity in cancer therapy rests on that fine balance point. Each and every cancer therapy targets some necessary and perfectly legitimate aspect of cellular function. The trick is to find targets that are absolutely essential for the survival of cancer cells, and use dosages where normal cells can still get by, still survive. The attractiveness of Hsp90 is that it is involved in the functioning of so many onco-proteins (proteins important in cancer cells). The downside is that Hsp90 is also involved in normal cell functioning, and therefore the potential risk of adverse effects if the dosage is too high or the drug is given for too long. Research in this area is definitely “work in progress”.
Some of the earlier broad spectrum review articles on 17-AAG come from the U.K. Two of the abstracts are given below.
Trends Mol Med. 2024 Feb;10(2):47-51.
Altered states: selectively drugging the Hsp90 cancer chaperone.
Workman P.
Cancer Research UK Centre for Cancer Therapeutics, Institute of Cancer Research, Sutton, Surrey, UK.
The molecular chaperone Hsp90 is an exciting cancer drug target. The first Hsp90 inhibitor to enter clinical trials--the geldanamycin derivative 17AAG--has recently demonstrated proof-of-concept for successful target modulation, with signs of therapeutic benefit. An important property of Hsp90 inhibitors is their ability to cause simultaneous, combinatorial blockade of multiple cancer-causing pathways by promoting the degradation of many oncogenic client proteins. However, the reason for therapeutic selectivity in cancer cells versus normal cells is unclear. New research now shows that Hsp90 exists in cancer cells in a heightened, activated state that is highly susceptible to inhibition by 17AAG.
PMID: 15106614
____________
Curr Cancer Drug Targets. 2024 Oct;3(5):297-300.
Overview: translating Hsp90 biology into Hsp90 drugs.
Workman P.
Cancer Research UK Centre for Cancer Therapeutics, Institute of Cancer Research, Sutton, Surrey SM2 5NG UK.
The Hsp90 molecular chaperone has emerged as one of the most exciting targets for cancer drug development. Hsp90 is over expressed in many malignancies, very likely as a result of the stress that is induced both by the hostile cancer microenvironment and also by the mutation and aberrant expression of oncoproteins. A particularly attractive feature of Hsp90 as a cancer drug target is that it is required for the conformational stability and function of a wide range of oncogenic 'client' proteins, including c-Raf-1, Cdk4, ErbB2, mutant p53, c-Met, Polo-1 and telomerase hTERT. Inhibition of Hsp90 should therefore block multiple mission critical oncogenic pathways in the cancer cell, leading to inhibition of all the hallmark traits of malignancy. This combinatorial blockade of oncogenic targets should give rise to board spectrum antitumour activity across multiple cancer types. The 'druggability' of Hsp90 was confirmed by the discovery that the natural products geldanamycin and radicicol, which have anticancer activity, exert their biological effects by inhibiting the essential ATPase activity associated with the N-terminal domain of the protein. The first-in-class Hsp90 inhibitor has entered clinical trial and provided proof of concept that Hsp90 can be inhibited and clinical benefit seen at non-toxic doses. Further development is underway and a related analogue 17DMAG also shows promise in preclinical models. In addition, novel Hsp90 inhibitors have been identified using methods such as high throughput screening and x-ray crystallography. The opportunities and challenges involved in translating the fast moving biology of Hsp90 into patient benefit is discussed.
PMID: 14529382
____________
Unfortunately, I have not been able to find too many papers describing the usefulness of 17-AAG specifically in CLL. Below is the only one I have come across thus far. It, too, is from British researchers, and describes cell studies using the parent compound geldanamycin, not its better tolerated offspring, 17-AAG. The sequence of cancer research goes something like this: cell line studies, followed by mouse studies, then larger animals such as dogs and monkeys, and finally in Phase I studies in humans. The paper below is interesting, but be aware that it talks only of cell lines in glass dishes, a far cry from human trials. The full paper is available free of charge, just click on the link given below. According to this paper, geldanamycin kills all B-cells. Healthy B-cells and cancerous CLL cells are killed with equal ease. In that regard it is similar to Rituxan, which targets CD20 expressed by all B-cells.
This paper reports that T-cells are more resistant to geldanamycin; not 100% safe, but they are a little harder to kill than B-cells. That is good news if it is indeed the case, and the margin of safety is high. The major problem with drugs such as fludarabine or Campath is that they kill not only all B-cells, they are also extremely toxic to T-cells. Loosing both B-cell and T-cell protection at the same time means deep immune suppression, which in turn opens the window of vulnerability to opportunistic infections, secondary cancers and so on. Drugs that do not target T-cells are safer, because they do not cause as deep an immune suppression. It is hoped that 17-AAG falls into the category of T-cell safe drugs. Some of the highlights from this article are summarized below:
Blood Journal Article (Free full-text)
Blood. 2024 Mar 1;103(5):1855-61. Epub 2024 Oct 23.
Geldanamycin and herbimycin A induce apoptotic killing of B chronic lymphocytic leukemia cells and augment the cells' sensitivity to cytotoxic drugs.
Jones DT, Addison E, North JM, Lowdell MW, Hoffbrand AV, Mehta AB, Ganeshaguru K, Folarin NI, Wickremasinghe RG.
Department of Hematology, Royal Free and University College Medical School, London, United Kingdom.
We studied the actions of geldanamycin (GA) and herbimycin A (HMA), inhibitors of the chaperone proteins Hsp90 and GRP94, on B chronic lymphocytic leukemia (CLL) cells in vitro. Both drugs induced apoptosis of the majority of CLL isolates studied. Whereas exposure to 4-hour pulses of 30 to 100 nM GA killed normal B lymphocytes and CLL cells with similar dose responses, T lymphocytes from healthy donors as well as those present in the CLL isolates were relatively resistant. GA, but not HMA, showed a modest cytoprotective effect toward CD34+ hematopoietic progenitors from normal bone marrow. The ability of bone marrow progenitors to form hematopoietic colonies was unaffected by pulse exposures to GA. Both GA and HMA synergized with chlorambucil and fludarabine in killing a subset of CLL isolates. GA- and HMA-induced apoptosis was preceded by the up-regulation of the stress-responsive chaperones Hsp70 and BiP. Both ansamycins also resulted in down-regulation of Akt protein kinase, a modulator of cell survival. The relative resistance of T lymphocytes and of CD34+ bone marrow progenitors to GA coupled with its ability to induce apoptosis following brief exposures and to synergize with cytotoxic drugs warrant further investigation of ansamycins as potential therapeutic agents in CLL.
PMID: 14576064
____________
Here's a nice PowerPoint presentation by Dr. Castro on HSP-90, 17-AAG and curbing the ZAP-70 signaling: J. C. Castro Presentation
You know me, I try to walk around and kick the tires a bit before I buy into things. This does not indicate any disrespect for the researchers coming up with cool science, just recognizes that patients put their lives on the line when they sign up for these clinical trials. I would like to see that everything possible is done to anticipate potential toxicity. If there are nasty surprises in store, they should be checked out as far as possible by appropriate mouse studies, as well as larger animal studies. Good clinical practice is a careful balance between protecting the human participants in clinical trials, while expanding the frontiers of our understanding. It concerns me that I have found so few papers on the use of 17-AAG specifically in CLL, nothing more than the cell line study above with the parent compound, and there is already talk of using this drug in live patients!
Below is one of the papers that caught my attention. It describes mouse and cell line study examining the use of 17AAG in breast cancer. Breast cancer often metastasizes to the bone marrow, a dangerous complication. This paper reports that use of 17-AAG increased this risk, and suggests that targeting Hsp90 may be contra-indicated for breast cancer patients. CLL is a different ball game, an entirely different kind of cancer. But heavy bone marrow involvement is an important negative prognostic indicator for us as well. Are there lessons to be learned here? I do not know.
Cancer Res. 2024 Jun 1;65(11):4929-38.
The heat shock protein 90 inhibitor, 17-allylamino-17-demethoxygeldanamycin, enhances osteoclast formation and potentiates bone metastasis of a human breast cancer cell line.
Price JT, Quinn JM, Sims NA, Vieusseux J, Waldeck K, Docherty SE, Myers D, Nakamura A, Waltham MC, Gillespie MT, Thompson EW.
Tumour Cell Migration and Metastasis Laboratory, St. Vincent's Institute, Melbourne, Vistoria, Australia.
Breast cancer metastasis to the bone occurs frequently, causing numerous complications including severe pain, fracture, hypercalcemia, and paralysis. Despite its prevalence and severity, few effective therapies exist. To address this, we examined whether the heat shock protein 90 (Hsp90) inhibitor, 17-allylamino-17-demethoxygeldanamycin (17-AAG), would be efficacious in inhibiting breast cancer metastasis to bone. Utilizing the human breast cancer subline, MDA-MB-231SA, previously in vivo selected for its enhanced ability to generate osteolytic bone lesions, we determined that 17-AAG potently inhibited its in vitro proliferation and migration. Moreover, 17-AAG significantly reduced MDA-MB-231SA tumor growth in the mammary-fat pad of nude mice. Despite these findings, 17-AAG enhanced the incidence of bone metastasis and osteolytic lesions following intracardiac inoculation in the nude mouse. Consistent with these findings, 17-AAG enhanced osteoclast formation 2- to 4-fold in mouse bone marrow/osteoblast cocultures, receptor activator of nuclear factor kappaB ligand (RANKL)-stimulated bone marrow, and RAW264.7 cell models of in vitro osteoclastogenesis. Moreover, the drug enhanced osteoclastogenesis in human cord blood progenitor cells, demonstrating that its effects were not limited to mouse models. In addition to 17-AAG, other Hsp90 inhibitors, such as radicicol and herbimycin A, also enhanced osteoclastogenesis. A pro-osteolytic action of 17-AAG independent of tumor presence was also determined in vivo, in which 17-AAG-treated tumor-naive mice had reduced trabecular bone volume with an associated increase in osteoclast number. Thus, HSP90 inhibitors can stimulate osteoclast formation, which may underlie the increased incidence of osteolysis and skeletal tumor incidence caused by 17-AAG in vivo. These data suggest an important contraindication to the Hsp90 targeted cancer therapy currently undergoing clinical trial.
PMID: 15930315
____________
There is no question about it, blocking the function of Hsp90 will make things uncomfortable for cancer cells on a number of fronts, and that is a good thing. Attacking the enemy on many fronts at the same time may make it harder for the cancer cell to escape death. The fact that cancer cells are more dependent on Hsp90 than regular cells gives us a potential window of opportunity, a way of killing the cancer cells without causing too much damage to the rest of the body. Ah, there is the rub. Exactly how much “collateral damage” do we expect? After all, Hsp90 is needed for the proper functioning of each and every cell in your body, not just the cancer cells. Could this become a case of cutting your nose off to spite your face? How do the risk and reward calculations stack up? In the next few weeks I will try to find more information on this topic, see if there are papers that I have missed in my search. It is entirely possible, and it would be good to find additional information. When I find out, you will be the first ones to know.
 Enter Keywords: |
———
Disclaimer: The content of this website is intended for information only and is NOT meant to be medical advice. Please be sure to consult and follow the advice of your doctors on all medical matters.
Copyright Notice:
Copyright © 2024-2007 CLL Topics, Inc. All Rights Reserved.
All materials contained on this site are protected by United States copyright law and may not be reproduced, distributed, transmitted, displayed, published or broadcast without the prior written permission of CLL Topics, Inc. You may not alter or remove any trademark, copyright or other notice from copies of the content.
However, you may download and print material from CLLTopics.org exclusively for your personal, noncommercial use.
———