 |
||||||||||
Date: November 7, 2026
by Chaya Venkat
Related Article:
Multi Drug Resistance to Chemotherapy.

Talk to any old-school oncologist, and you will probably get the advice that unlike solid cancers such as breast cancer, colon cancer, etc., the best thing to do with CLL is to "Watch and Wait". You will get very little argument from the experts that earlier diagnosis translates into far better prognosis for most solid cancers, since therapy can be initiated that much sooner. But in the case of CLL, conventional wisdom is headed the other way around. What is the advantage of earlier diagnosis of CLL if there is nothing to do? Why have several years more of "Worry & Wait", would it not be better to find out as late as possible, just in time to initiate chemotherapy? I don't know about you folks, but it would drive me nuts not to know. I suspect that is true for most of us Type A personalities. Heck, I have paid for the apple from the tree of knowledge, look at all my tax dollars that go to support medical research, why would I not want to get a bite of this expensive apple?
Putting that aside, is there any real advantage in making analysis of genomic aberrations in CLL patients a standard, necessary for good clinical practice? If you have read two recent articles on this website, What Type of CLL Do You Have? and the article review, Prognosis at Diagnosis, you already know the answer. The old study done with chlorambucil that showed no survival advantage in early initiation of therapy is outdated and no longer valid logic for making therapy choices today. Now we have many more treatment options, many more drugs available to us. Knowing a bit more about the type of CLL you have, and catching it earlier, may give you low-toxicity therapy options that may not be effective later on. In the opinion of this lay person observer, Watch & Wait as standard strategy for each and every newly diagnosed CLL patient does not work any more. I am sure there is a version of Murphy's Law that says all things change over time, and in true Murphy style, when things change in the course of progressing CLL, they always change for the worse. This article and the next one in this series try to outline some of the changes that take place as CLL progresses.
You may have started with one of the more benign chromosomal aberrations, but that does not mean it will stay that way for ever. True, for some lucky percentage of patients with a good phenotype, things stay pretty much constant over their entire lives, their CLL never progresses and they are the true "smoldering CLL" variety. But in a substantial subset of patients, new, and often more dangerous, mutations accumulate over time. These new variants of the original clone may be better suited to survive and thrive, and pretty soon they become the dominant species. This is Darwinian selection at a cellular level, survival of the fittest clone. You don't need Murphy's law to tell you that CLL clones with enhanced survival capabilities are that much harder to kill.
As you can see from the first abstract below, when patients were monitored for five years, clonal evolution was seen to happen quite frequently. In fact, there is growing consensus that disease progression is precipitated by the emergence of a new and more virulent clone. The second abstract gets into more detail. 82 patients were followed for about 45 months. Over that period of time, some of these patients developed several new karyotype mutations that they did not have at the start of the study. In the order of increasing risk factor, 4 patients developed 13q mutation, 3 patients developed 12trisomy, 8 patients developed 11q mutation, one patient developed the poor prognosis 17p mutation, and one developed the relatively rare and poor prognosis 6q deletion. Invariably, disease progression kept pace with acquisition of new chromosomal abnormalities.
The third abstract confirms these findings. Not only does clonal evolution take place in CLL, it is quite common. In little over 4 years, fully 41% of all patients had added new chromosomal aberrations to their repertoire, aberrations that they did not have to begin with. In this study too, there was a close match between disease progression and the appearance of a new and more dangerous clone. What does this say about the therapy strategies? Well, it seems to me that not only is it important to know what type of CLL you have, it is also important to verify periodically that you still have what you thought you had, and your game plan is still valid for the version you now have.
In case it has slipped your mind, let me emphasize the importance of the particular chromosomal aberration you have. By now you are probably aware of the importance of Rituxan as a relatively low toxicity drug in the treatment of B-cell malignancies. Let us consider the patients who developed new 11q and 17p deletions over the course of 4 years in the studies described above. At the beginning of their journey, they would have been in "Bucket A". If they had stayed in the traditional Watch & Wait mode for four years, at the end of that period they would most likely be in higher Rai stage, with worse chromosomal aberrations, heavier tumor load, possibly spleen and liver involvement. We do not have information about their B2M CD38 level, but there have been reports of CD38 changing over time, and certainly B2M increases with advanced disease. All in all, chances are very good that the patients would be in "Bucket C" after the 4-year Watch & Wait period.
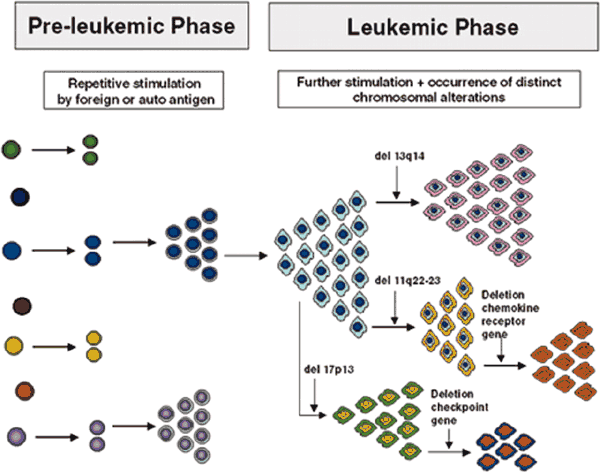
Model for the Development and Evolution of B-CLL Cells ASH Education Book 2026:
Biology and Treatment of Chronic Lymphocytic Leukemia Note that the specific chromosomal changes are illustrations of the principle of clonal diversification; their presence and sequence of occurrence are not to be taken literally. |
Why is this important? By now we all know that Rituxan is an important monoclonal antibody in the treatment of CLL. When used as frontline and single agent therapy, Rituxan provides a low toxicity approach to treating CLL. Neither it nor its sister monoclonal Campath do a very good job on bulky lymph nodes. Even if that were not the case, the fourth abstract below gives one important reason to consider clonal evolution when deciding your long term strategy. There seems to be strong reason to believe that patients with 17p and 11q deletions do not respond as well to Rituxan. I do not know if this would translate into poorer response, as well, to combination chemotherapies using Rituxan, such as the FRC and RF protocols. If that were the case, these patients may have been better off using Rituxan (or its combinations with other chemotherapy drugs) earlier in their disease cycle, while they still had relatively benign chromosomal aberrations. 17p deletion is not good news, patients with this particular chromosomal aberration do not respond well to many drugs.
Does the chance of developing a new variant with different and more dangerous chromosomal aberration increase with increasing number of CLL cells in your body? I would think this would be a matter of statistics and probability. Look at it this way: when you buy a single lottery ticket, your chances of winning the jackpot are very slim, one in many million. Most of us have contributed to "lottery pools" at work over our careers, where a bunch of people get together, pool their money and buy a whole bunch of lottery tickets. This is still no guarantee that the "pool" will win the jackpot, but their chances are better than winning with a single ticket. Developing 17p deletion is the equivalent of winning the jackpot, in a bad sense. My guess would be that the statistical chances of developing this mutation in a given patient would be lower with a smaller number of CLL cells that could initiate this change, than if the same patient had many, many more CLL cells that could potentially do so.
Does this mean I am recommending shoot first and ask questions later, treat each and every CLL patient as soon as they are diagnosed? Heck no, that too would be a "one size fits all" approach, just like conventional Watch & Wait. Jumping the gun and over treating an indolent disease is as foolish as letting a more aggressive form simmer to the point where it is hard to control. You might enjoy reading the case history of two hypothetical patients Jane and John, where this specific issue is highlighted (ZAP-70: Why Should You Care?). I admit the scenarios painted are just a little over the top (but not by much), to get across the point I was trying to make. I do not recommend therapy choices, but I do suggest that you discuss with your doctors the specifics of your case, your specific prognostic information, and your therapeutic options as an unique individual. Tailoring customized therapy will also take into account the patient's life style, personality and support systems.
Combining the information regarding clonal evolution presented in this article with the "Bucket theory of CLL" defined earlier, I have developed two flow-charts that compare the two approaches to treatment.
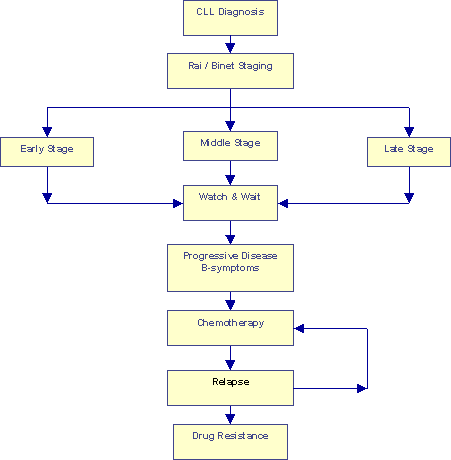
Conventional "Watch and Wait" Approach |
The flow chart above describes the conventional "Watch & Wait" approach. True, patients are segregated into different groups based on their Rai and Binet classifications. But nothing much seems to be done with that segregation, every one goes through the Watch & Wait period until the onset of B-symptoms. Chemotherapy is initiated at that point. Remissions, even the so-called "Complete Responses" or CRs, do not last forever. Eventually all patients relapse. Another sad but real fact of life is that most patients develop multiple drug resistance, sooner or later. The end game in this approach is a series of increasingly aggressive chemotherapy regimes with progressively shorter remissions in between, perhaps a bone marrow transplant.
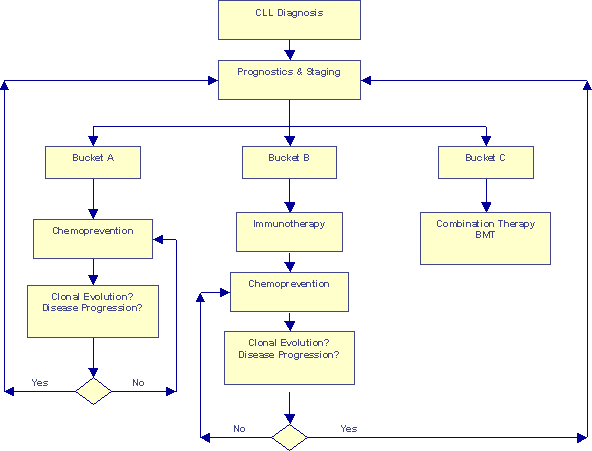
Prognosis-based Risk-adjusted Approach |
This second flow-chart represents some of the newer concepts we have discussed on Topics. We have discussed segregating patients into three risk categories based on a detailed analysis of chromosomal aberrations and other prognostic information. The earlier article What Type of CLL Do You Have? suggests customized game plans for the different risk categories. This flow chart goes one step further, it make room for periodic re-evaluation of patients, to make sure they have not had a clonal evolution event that would kick them into the next Bucket, requiring a new strategy for optimum benefit.
I have used the word "chemoprevention" on this chart to mean a whole group of approaches:
Most of us would rather the "cure for cancer" came in the form of a nice little pill, a whole lot easier than the hard work of modifying our own actions and lifestyle choices. We would also like to blame someone or something else for the increasing number of people with heart disease and cancer. It must be because of pesticides, pollution, cell phones, microwave ovens, aliens from outer space, or some dark conspiracy of the medical and pharmaceutical establishments, aided and abetted by the FDA and the NCI. I am sure pollution, pesticides and the like have a role to play, but an even bigger share of the blame lies with our own life style choices. Statistically speaking, obesity, smoking, poor nutrition and lack of exercise are probably the biggest culprits in heart disease, diabetes and cancer in America.
Similarly, I used the word "immunotherapy" on this chart to mean a whole group of therapeutic approaches such as monoclonal antibodies (Rituxan and Campath); immune modulating drugs such as Leukine, Neupogen, PT-100, IL-2, Interferon, glucosteroids; newer approaches such as ex-vivo grown CTL therapy, bispecific antibodies, "gene" therapies, heat-shock vaccines, idiotype vaccines, DNA vaccines, etc. I am sure I have missed at least a few from my list.
For the majority of us the day is not here yet, when we can sit back secure in the knowledge that our doctors will develop the perfect fit therapy choices customized for our unique individual needs, without any involvement or input from us. This may be the case for a lucky few patients. For the rest of us, knowledge gained by hard work is an essential part of empowerment; so too a sense of community with fellow patients. Working together we may be able to influence public health policy decisions more than each one of us can do as single patients.
Leukemia. 1995 Dec;9(12):2003-8.
Karyotypic evolution in CLL: identification of a new sub-group of patients with deletions of 11q and advanced or progressive disease.
Fegan C, Robinson H, Thompson P, Whittaker JA, White D.
Department of Haematology, University Hospital of Wales, Heath Park, Cardiff, UK.
Chronic lymphatic leukaemia (CLL) is the most common leukaemia and is characterized by long-term survival. Previous studies have shown that karyotypic abnormalities are relatively stable and that certain abnormalities may be associated with a poor prognosis. In a prospective 5-year study of 45 patients with typical CLL, sequential karyotypic studies were undertaken every 6-12 months. Clonal karyotypic abnormalities were identified in 62% of patients, either at diagnosis or during the study period with 38% (17/45) exhibiting clonal evolution. In patients with no clinical disease progression, 13q abnormalities were most commonly detected compared with 11q deletions in patients with progressive disease. Karyotypic evolution was significantly associated with progressive disease (12/16, 75% vs 5/29, 17%; P < 0.001, chi2). Thus, karyotypic evolution is not uncommon in CLL, is usually associated with disease progression and deletions of 11q are the commonest detected abnormalities.
PMID: 8609709
____________
Haematologica. 2026 Jan;87(1):44-51.
Late appearance of the 11q22.3-23.1 deletion involving the ATM locus in B-cell chronic lymphocytic leukemia and related disorders. Clinico-biological significance.
Cuneo A, Bigoni R, Rigolin GM, Roberti MG, Bardi A, Cavazzini F, Milani R, Minotto C, Tieghi A, Della Porta M, Agostini P, Tammiso E, Negrini M, Castoldi G.
Institute of Hematology, University of Ferrara, via Savonarola 9, 44100 Ferrara, Italy.
BACKGROUND AND OBJECTIVES: Chromosome 11q22.3-23.1 deletions involving the ataxia-teleangiectasia mutated (ATM) locus (11q-/ATM+/-) are detected at diagnosis in 10-20% of cases of B-cell chronic lymphocytic leukemia (CLL) and are associated with a relatively aggressive disease. The aim of this study was to ascertain whether 11q-/ATM+/- may appear late during the course of the disease and to analyze its possible correlation with disease evolution.
DESIGN AND METHODS: Eighty-two patients with CLL and related disorders, i.e. CLL/PL and prolymphocytic leukemia (PLL), without 11q- at diagnosis were sequentially ascertained at 1-2 year intervals by conventional cytogenetic analysis (CCA) and fluorescence in situ hybridization (FISH), using an ATM-specific probe.
RESULTS: Eight patients acquired a submicroscopic 11q deletion 13-43 months after diagnosis: the diagnosis at presentation was CLL in 3 cases, CLL/PL in 3 cases and PLL in 2 cases. A 13q14 deletion preceded the development of 11q- in four patients; additional aberrations included +12 (three cases), 17p13 deletion and 6q21 deletion (one case each). The acquisition of the 11q deletion was more frequently found in those patients presenting with CLL/PL and PLL than typical CLL (p=0.0016) and with splenomegaly (p=0.003). Follow-up data showed that karyotype evolution (p=0.009) and cytological transformation (p<0.001) were associated with the acquisition of this cytogenetic lesion. The variables predicting for a shorter survival in this series included the 11q deletion (p=0.03), along with other classical clinicobiological parameters (performance status, advanced stage, splenomegaly, elevated serum beta2 microglobulin and lactate dehydrogenase levels.
INTERPRETATION AND CONCLUSIONS: a) Submicroscopic 11q deletion involving the ATM locus may, in some instances, represent a secondary change in CLL, CLL/P and PLL, suggesting that sequential FISH analyses are necessary to detect this chromosome anomaly in some patients; b) the acquisition of 11q-/ATM deletion may play a role in determining cytological transformation and disease progression of CLL and related disorders.
PMID: 11801464
____________
Am J Hematol. 1998 Nov;59(3):223-9.
Secondary abnormalities of chromosome 6q in B-cell chronic lymphocytic leukemia: a sequential study of karyotypic instability in 51 patients.
Finn WG, Kay NE, Kroft SH, Church S, Peterson LC.
Department of Pathology, Northwestern University Medical School, Chicago, IL
Although karyotypic abnormalities are well documented in B-cell chronic lymphocytic leukemia (B-CLL), few sequential cytogenetic studies have been done. In this study, peripheral blood lymphocytes from fifty-one patients with B-CLL were sequentially karyotyped over a mean interval of 13.8 months (range, one to 51 months). Cytogenetic clones were detected in 33/51 patients (66%) on initial study, including 17 patients with structural abnormalities of chromosome 13q14, and three patients with trisomy 12. Karyotypic evolution was documented in 22/51 patients (43%). The most common secondarily acquired chromosome aberrations were structural abnormalities of the long arm of chromosome 6 involving the region of 6q21-q24 (six patients). Four patients each had acquired structural abnormalities of 1q, 3p, 12q, and 13q. Disease progression, as measured by advance in Rai stage or death from the disease, was observed more often in the clonal evolution group than in the karyotypically stable group (11/22 vs. 5/29; P = 0.017). Patients with secondary abnormalities of 6q had a significantly decreased progression-free survival interval compared with other patients in the study (P = .023). The authors conclude that clonal karyotypic evolution is common in B-CLL, and that clonal evolution correlates with clinical disease progression. Furthermore, the poor outcomes previously attributed to CLL with 6q abnormalities may be related to the clonal acquisition of these abnormalities over time. Future studies should focus on the relevant genetic events underlying the clinical progression observed with karyotypic evolution of B-CLL.
PMID: 9798660
____________
Cancer Res. 2026 Jan 1;63(1):36-8.
Interphase cytogenetic abnormalities in chronic lymphocytic leukemia may predict response to rituximab.
Byrd JC, Smith L, Hackbarth ML, Flinn IW, Young D, Proffitt JH, Heerema NA.
The Division of Hematology-Oncology, Ohio State University, Columbus, OH
Select cytogenetic abnormalities such as del(17)(p13.1) and del(11)(q22-q23)predict rapid disease progression and inferior survival in chronic lymphocytic leukemia (CLL). We sought to determine the impact of the four most common interphase cytogenetic abnormalities in 28 CLL patients relative to response to three-times-a-week rituximab therapy. Abnormalities were noted in 25 of the 28 patients to include del(13)(q14.3) [n = 16 (57%)], del(11)(q22.3) [n = 10 (36%)], +12 [n = 6 (21%)], del(17)(p13.1) [n = 5 (18%)], and normal [n = 3 (11%)]. Only a minority of each of these occurred as sole abnormalities. To categorize patients into one specific group, we used the hierarchical order del(17)(p13.1) > del(11)(q22.3) > trisomy 12 > del(13)(q14.3) to prioritize. Response to rituximab was noted to vary by cytogenetic group: del(17)(p13.1), 0% [n = 5]; del(11)(q22.3), 66% [n = 9]; del(13)(q14.3), 86% [n = 7]; +12, 25% [n = 4], and normal, 0% [n = 3]. Response was significantly lower (P = 0.05) in patients with del(17)(p13.1) as compared with those with other abnormalities. These data suggest that interphase cytogenetics in CLL may be predictive of a response to rituximab therapy and provide support for additional studies validating risk-adapted therapy in this disease.
PMID: 12517774
____________
 Enter Keywords: |
———
Disclaimer: The content of this website is intended for information only and is NOT meant to be medical advice. Please be sure to consult and follow the advice of your doctors on all medical matters.
Copyright Notice:
Copyright © 2026-2007 CLL Topics, Inc. All Rights Reserved.
All materials contained on this site are protected by United States copyright law and may not be reproduced, distributed, transmitted, displayed, published or broadcast without the prior written permission of CLL Topics, Inc. You may not alter or remove any trademark, copyright or other notice from copies of the content.
However, you may download and print material from CLLTopics.org exclusively for your personal, noncommercial use.
———




