 |
||||||||||
Date: January 1, 2026
by Chaya Venkat

If there is a single most important fact you need to know about CLL right upfront, this is it: one shoe does not fit all. CLL comes in many flavors and sizes. The spectrum stretches all the way from a “good” cancer that is not going to do much of anything for the rest of your normal life span to an aggressive, incurable cancer that requires treatment soon after diagnosis, compromise your quality of life with frequent infections and ultimately kills you in a few short years.
Just a few years ago we had no way of knowing which risk bucket you were in, we had no way of predicting with reasonable accuracy your likely prognosis. Now we do. We have modern prognostic testing that will give you a good view of the lay of the land, what you can expect down the road. Just in case you are toying with the head-in-sand strategy as a way of dealing with your CLL diagnosis, let me point out that smart therapy choices made by you and your oncologist can make a huge difference in both the quality and quantity of your life. What you don’t know about CLL can literally kill you a lot sooner than otherwise. How is that for motivating you to find out more about your particular brand of CLL?
A 2008 article in the prestigious industry journal “Blood” looked at CLL patient profile (USA) over two decades over the time period of 1973 – 2026.
| Calendar Period | ||||||
| Age/Sex | 1980-84 | 1985-89 | 1990-94 | 1995-99 | 2000-04 | Total |
| All | 3,642 | 3,968 | 4,236 | 4,191 | 4,454 | 20,491 |
| 15-59 | 670 | 762 | 812 | 928 | 965 | 4,137 |
| 60-69 | 1,079 | 1,115 | 1,164 | 956 | 1,055 | 5,369 |
| 70-79 | 1,096 | 1,252 | 1,311 | 1,343 | 1,303 | 6,305 |
| 80+ | 797 | 839 | 949 | 964 | 1,131 | 4,680 |
| Men | 2,177 | 2,355 | 2,451 | 2,492 | 2,645 | 12,120 |
| Women | 1,465 | 1,613 | 1,785 | 1,699 | 1,809 | 8,371 |
In a word, yes. And they are enjoying better quality of life.
The good news is that with better understanding we are able to classify patients into distinct risk profiles that need different management strategies. "Management" of CLL is an important concept. I would not advise you to expect overnight miracles, a newly discovered magic pill that will cure your CLL once and for all. That is not how medical research works. But in the life time of patients reading this article, "management" of this disease is very much in the cards for a large majority and that is nothing to sneeze at.
If you do not think much of the idea of "management" of CLL, think of all the patients with high blood pressure, diabetes and other chronic diseases. With proper diagnosis, medical care and "management" of their diseases, their quality of life and overall survival have improved dramatically from just a decade ago. They learn to live with their diseases, learn to manage the symptoms and drug therapies while they continue to live productive lives as parents, spouses, tax-payers and good citizens. They do not get the pitying glances that most of us have learned to hate, one of the side effects of a cancer diagnosis.
You dont have to take my word for it that our guys are living longer than they used to. Here is a graph that shows overall survival of CLL patients, depending on when they were first diagnosed. 50% of patients diagnosed between 1980 and 1984 did not make it past 7.5 years. But patients diagnosed ten years later, between 1990 and 1994, half of them were still around at 12 years. That is a solid gain, and my guess is that the improvement would be even better when we look at patients diagnosed another decade later. Just look at the rate at which our understanding of this disease is growing! Dont you think it would be a shame if you were diagnosed recently but got stuck with 1980 survival odds because of lack of modern understanding of this disease?
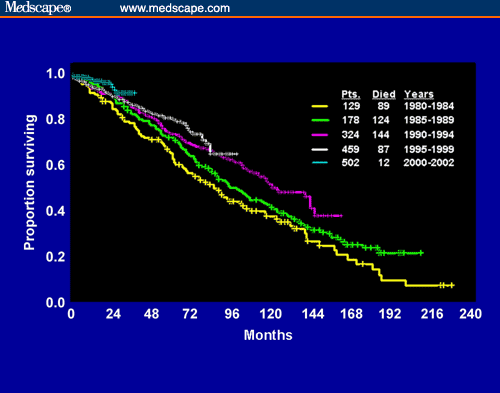
Source: Management Strategies for Chronic Lymphocytic Leukemia by Michael J. Keating.
http://www.medscape.com/viewarticle/456068
Most CLL experts would agree that CLL patients fall into three roughly equal size risk categories. We coined the term Risk Buckets to describe them.
For a third or so of "lucky" CLL patients diagnosed recently, lucky to have good prognostic indicators, access to good medical care and physicians that understand this disease, CLL may soon become nothing more than a nightmare that they can put behind them. These guys are often said to have a smoldering variety of CLL. They will continue to have CLL but their disease grows so slowly that for all practical purposes it will have no impact on their lives. It may be a long time before they need any therapy to control their CLL. In fact, rushing to treat smoldering CLL patients with aggressive chemotherapy protocols is both foolish and dangerous. Would you take a big stick and beat a hornets nest that is slumbering peacefully and doing no harm to anyone? Watch & Wait for as long as the disease is behaving itself is the best thing to do with smoldering CLL. Chances are good that lucky patients in this low risk category (Bucket A) will die in their beds of ripe old age some day, or get hit by the proverbial bus with their name on it or whatever, none of it having anything to do with their CLL.
For next third of patients CLL may be "managed" to the point where its impact on their lives is reduced and they can look forward to good quality of life for many years ahead. This is the middle risk Bucket B. For elderly patients newly diagnosed with this middle-of-the-road variety of CLL the impact of the diagnosis on their life expectancy will be minor. A seven or eight year survival projection for a patient who is in his middle seventies at the time of diagnosis may not be all that terrible. But for younger patients the same 7-8 year survival prognosis can be a devastating blow. I know this, up close and personal. My husband was 52 when he was diagnosed. He was only 59 when he died, seven short years later. Reality check: CLL can and does kill patients in high risk categories.
For the last third of patients with the most aggressive form of CLL and all the bad prognostic indicators, accurate diagnosis and risk classification are even more important. These folks are in high risk Bucket C. They don't have the luxury of a lot of time to waste. Dithering is not an option for them. For this unfortunate subset of CLL patients, making the right first therapy choices is essential. Under-treating an aggressive form of CLL, waiting too long or wasting time with ineffective and tentative therapies spells trouble of the worst sort.
If you are a younger patient with high risk profile, the game plan becomes a lot more critical. Unlike more elderly patients with a middle of the road prognosis, you are not going to be able to run out the clock. A five to ten year survival prognosis is not good enough, you face too much of a penalty in reduced life span. Even our best chemotherapy regimens are not going to hold the line for what would otherwise be your normal life span. I am extremely happy to report that modern advances in stem cell transplants now give us options we did not have just a few years ago. Emerging consensus is that young patients with high risk CLL should be looking at stem cell transplants sooner rather than later.
Think of stem cell transplants as getting rid of your cancerous, no-good-bum of an immune system and replacing it with a healthy immune system from a compatible and willing donor. It helps if you have a well matched sibling donor, since it saves the hassle of finding a matched unrelated donor, a task that becomes next to impossible for ethnic minorities. Stem cell transplants are coming up the curve awfully fast. Survival statistics are improving each year. But making the decision to go the transplant route is still a tough call and you really need to get your ducks in a row before you can make it. We have a lot of information about transplants on CLL Topics and CLL Topics Updates that will help you understand the risks and rewards.
You have made the first step: you now understand CLL comes in three roughly equal risk categories. If you are like most newly diagnosed patients you want to compare your own test results against prognostic yard-sticks and figure out which risk "bucket" you belong to. Bear in mind, the buckets do slosh and splash and some one in a low risk bucket may "graduate" and move up the scale to the next bucket over time. All the same, getting a reality check of your own prognosis at the time of diagnosis is an important thing to do. How can you fight an enemy you do not understand?
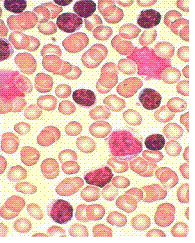 CLL is a blood cancer. It is a cancer of B-cells, a type of white blood cells that circulate all through your body wherever blood circulates. Alongside is a picture of what CLL cells look like when they are viewed under a microscope. CLL cells are small, round B-cells (stained dark purple in this slide). Often, there are smudge cells; think of them as cells that have gotten squished flat. It seems CLL cells are a bit more fragile than normal B-cells and they are more likely to get bent out of shape in the process of making a microscope slide of the sample. You can see some of these smudge cells in the picture. This is the picture the pathologist would see under the microscope. If the lab report mentions smudge cells, you now know what that is all about.
CLL is a blood cancer. It is a cancer of B-cells, a type of white blood cells that circulate all through your body wherever blood circulates. Alongside is a picture of what CLL cells look like when they are viewed under a microscope. CLL cells are small, round B-cells (stained dark purple in this slide). Often, there are smudge cells; think of them as cells that have gotten squished flat. It seems CLL cells are a bit more fragile than normal B-cells and they are more likely to get bent out of shape in the process of making a microscope slide of the sample. You can see some of these smudge cells in the picture. This is the picture the pathologist would see under the microscope. If the lab report mentions smudge cells, you now know what that is all about.
Chances are that this whole thing started when a routine blood test showed you have elevated White Blood cell Count (WBC), and more specifically you had high Absolute Lymphocyte Count (ALC). (Please read our recent articles on CBC, ALC, red blood cells and platelets to become familiar with these acronyms).
CLL diagnosis requires ALC to be higher than 5,000. But an elevated ALC and elevated WBC are not by themselves sufficient to confirm CLL. Heck, healthy folks with no cancer of any kind have elevated WBC and ALC when they come down with an infection of some sort! This is perfectly normal and healthy reaction. Our bodies gear up for a fight by increasing the troop numbers. White blood cells (which include B-cells, lymphocytes) are the defenders of our bodies; they are the frontline troops that get mobilized when an invading pathogen is detected. In healthy folks elevated WBC and ALC go back down to normal levels once the infection has been resolved.
OK, you have elevated WBC and ALC that refused to go away even after you dutifully took the broad spectrum antibiotic course your GP prescribed. Looks like what you have is not a simple infection. The plot thickens, your GP probably sent you to the oncologist at this point to check things out.
The next step is to conduct a flow cytometry test. This too is a blood test and most testing labs these days can handle it without fuss.
All cells in your body have a number of special structures on their surfaces. These are given names cluster designations or CD numbers. Different cell types have different set of CD types present on their surface. All members of a given family of cells have the same set of CDs, a sort of family fingerprint.
CLL is a cancer of B-cells, one kind of white blood cell. CLL cells have their own particular profile of CDs. Think of it as a fingerprint of this particular variety of cancer cells. The fingerprint is different if we are talking of some other B-cell cancer. Flow cytometry looks for this unique fingerprint and it is very useful in confirming CLL as well as ruling out some of the kissing cousin cancers. It is particularly important to rule of more dangerous varieties of B-cell cancers such as mantle cell lymphoma.
The table below gives the fingerprint profiles of the various B-cell cancers. Depending upon the sophistication of your testing lab all or only some of these CDs may be studied and reported. Flow cytometry looks not only to see what CDs are present, but also how densely they are packed on the cell. For example, bright expression of a particular CD type (say, CD19) means there are many thousands of copies of CD19 present on each cell. A dim expression of CD20 for example means there are far smaller numbers of CD20 groups present per CLL cell. Some labs use + for dim, ++ for intermediate and +++ for bright expression of various CD clusters.
| Cancer Type | Sig | CD5 | CD19 | CD20 | CD10 | CD11c | CD22 | CD23 | CD43 | CD103 | Cyclin D1 |
| CLL/SLL | + | + | + | + | - | - | - | + | + | - | - |
| MCL | ++ | + | + | + | +/- | - | + | - | + | - | + |
| HCL | ++ | - | + | + | - | + | + | - | - | + | |
| LPL | ++ | - | + | + | - | - | + | - | - | - | - |
| SML | ++ | - | + | + | - | - | + | - | - | - | - |
| FCL | ++ | - | + | + | + | - | + | - | - | - | - | Abbreviations: CLL, chronic lymphocytic leukemia; SLL, small lymphocytic lymphoma; MCL, mantle cell lymphoma; HCL, hair cell leukemia; LPL, lymphoplasmacytic lymphoma; SML, splenic marginal zone lymphoma; FCL, follicular cell lymphoma. |
In the vast majority of cases, a complete CBC with non-trivial elevation in WBC and ALC followed up by a flow cytometry to confirm CLL phenotype (molecular fingerprint with all the right CDs present and none of the wrong ones present) – this is sufficient to confirm diagnosis of CLL. Now that we have that sorted out and we are all CLL patients here, and the odd SLVL or MCL folks tagging along thus far have logged off, let us get on with the job of defining the three buckets.
Every cancer starts with a single cell that has some defect that makes it cancerous. It becomes a problem when it passes along the same defect to all its daughters, granddaughters etc, multiplying into great hordes of cancerous cells. In the case of CLL, five distinct commonly recurring chromosomal defects have been identified. FISH analysis has demonstrated that more than 92% of patients can be grouped into five groups, based on their particular chromosomal aberration. The table below lists the abnormalities, in the order of decreasing risk.
17p deletion is considered the most dangerous abnormality, since it is often accompanied by deletion of the tumor suppressor gene p53. At the other end of the risk spectrum, people with 13q deletion as their sole genetic abnormality are likely to live out their full life spans, and not have to deal too much with CLL.
One patient can have more than one type of aberration. If your FISH report cites more than one chromosomal defect, I guess I would then look at the "worst" aberration as the one in charge of driving your CLL bus. If you would like to know more about chromosomal defects and what they mean in blood cancers, go to the article titled "Genetic Abnormalities in Blood Cancers".
| Karyotype* | No. of Patients | % of Total |
| 17p deletion | 23 | 7 |
| 11q deletion | 56 | 17 |
| 12q trisomy | 47 | 14 |
| Normal Karyotype | 57 | 18 |
| 13q deletion as sole abnormality | 117 | 36 |
| Various abnormalities | 25 | 8 |
* The five major categories are defined a follows: |
||
Source: Genomic Aberrations and Survival in CLL
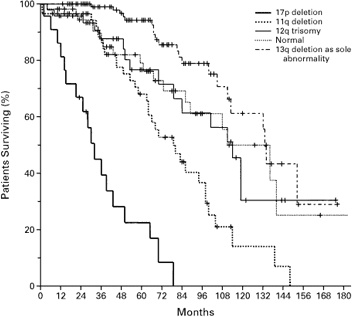
Probability of Survival from the Date of Diagnosis
for the Five Genetic Categories
Dohner, et al., NEJM
The median survival times (the time by which point half of the patients are alive, the other half dead) for the groups with 17p deletion, 11q deletion, 12q trisomy, normal karyotype, and 13q deletion as the sole abnormality were 32, 79, 114, 111, and 133 months, respectively. That is quite a difference between the different groups, all the way from less then 3 years for the unlucky 17p deleted cases to just under 10 years for the smoldering 13q deleted variety.
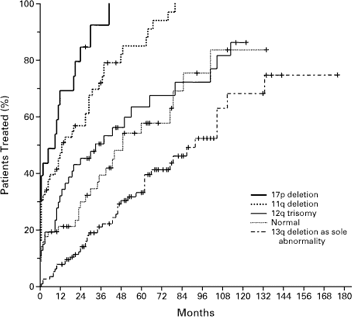
Probability of Disease Progression, as Indicated by the
Treatment-free
Interval in the Five Genetic Categories
Dohner, et al., NEJM
The median treatment-free intervals (the time it took for half the group to need therapy, starting the clock when they were first diagnosed) for the groups with 17p deletion, 11q deletion, 12q trisomy, normal karyotype, and 13q deletion as the sole abnormality were 9, 13, 33, 49, and 92 months, respectively. The aggressive 17p deleted cases needed treatment almost right away, an average of 9 months between when they were diagnosed and when they needed treatment. At the other extreme, the 13q deleted folks could coast along for more than seven years before they needed any kind of treatment. Quite a difference, you will admit. This is why prognostic testing is important. It gives you a chance to figure out what is coming at you down the road, a chance to come up with a sensible game plan that works for you and your family.
The single most important bit of information a CLL patient can have right up front is whether or not he has the dreaded 17p deletion reported in his FISH test report. This chromosomal defect influences the rate at which the CLL grows as well as resistance of the cancer to most commonly available therapies. The following graph highlights the effect of a p53 deletion upon the prognosis for a CLL patient. There is a huge difference in the survival patterns of patients, depending on whether or not they have this important chromosomal defect.
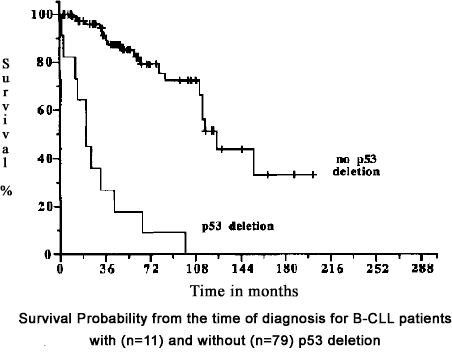
Effect of p53 gene deletion on survival in CLL
Dohner, et al., Blood.
Link: Blood Journal: p53 Gene Deletion in Chronic B Cell Leukemias
But it is important to remember this point: even if fate has dealt you a poor hand, the idea is to play it to best effect. Even patients with so-called poor prognosis can do very well, provided they take charge of the situation and avoid the trap of the head-in-sand syndrome. What these guys cannot afford is bad luck combined with bad medical care.
As you read this and other sections in this long article, bear in mind this very important fact: all the survival data presented in this essay are based on historical data. As we discussed right at the top of this article, CLL landscape is changing for the better and patients diagnosed more recently can hope to do better than the historical norms. It is also important to understand that statistical information based on large groups of patients gives us a heads-up on what to expect, gives us the odds as it were. It does not guarantee how things will play out in your individual case. You may turn out to be luckier than the general run of the mill patient in your risk bucket. Or not.
FISH analysis of chromosomal abnormalities is an incredibly important and powerful tool. It is a shame that it is not used more routinely. If you go to one of the expert cancer centers FISH testing is routinely done. Patients with 13q deletion as the sole abnormality are in the low risk Bucket A. Patients with 12 trisomy (as well as those patients who have none of the four commonly monitored abnormalities and often classified as normal) are in the middle Bucket B. Patients with 11q and 17p deletions are in the high risk Bucket C.
After getting your FISH results it would be nice if you can go home reasonably clear about your risk profile for the rest of your CLL career. Unfortunately, things do change over time in CLL. Clonal evolution happens frequently in CLL. As time goes on for a subset of patients, their CLL clone acquires more genetic abnormalities, becoming more and more bizarre. Over a period of about 4 years, the authors of a recent study detected additional abnormalities acquired by 22 out of 51 patients. Disease progression, and / or death from the disease was observed more often in the clonal evolution group than in the chromosomally stable group. The authors concluded that clonal evolution is common in B-CLL, and that clonal evolution correlates with clinical disease progression. Bottom line, it makes sense to check your FISH status periodically to confirm that you are still in the bucket you thought you were in and therefore your game plan is still valid for your situation. It is particularly appropriate to get re-FISHed before you start on a new therapy regimen, to make sure the therapy is appropriate in your case.
This has been an important prognostic indicator and fortunately for CLL patients, it is one that can be measured routinely by commercial testing labs using just a blood sample. You may not want to get it done as frequently as your CBC, but it is certainly worth getting it done every so often - especially if you and your oncologist feel something unusual is happening re your CLL. Some expert centers swear by this prognostic indicator (such as M. D. Anderson, Houston TX).
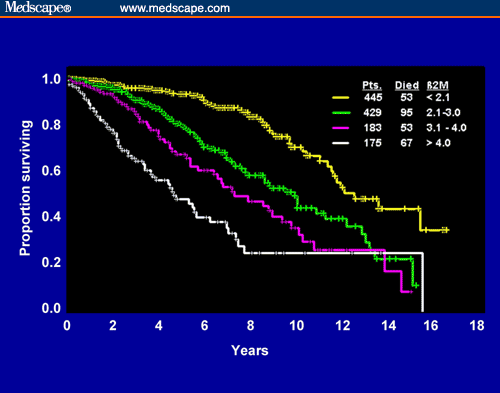
Relationship of β2M to survival in CLL
Source: Management Strategies for Chronic Lymphocytic Leukemia by Michael J. Keating.
With data from more than 1,200 people, M. D. Anderson has developed survival statistics based on β2M levels. As you can see from the graph here, we can reasonably divide patients into our three buckets: patients with β2M less than 2.0 are in Bucket A; patients with β2M between 2.0 and 4.0 are in Bucket B; patients with β2M greater than 4 are in Bucket C. The cut-off points are obviously arbitrary, and β2M levels also change over time, especially if you are going through therapy. That is why it is a good idea to get repeat testing of B2M at regular intervals.
This prognostic indicator has revolutionized our ability to define patients into low risk and high risk categories. You can learn a lot more of the details of what this is all about in other articles on this website: IgVH Gene Mutation Status and CD38 Expression and ZAP-70 and IgVH Gene Mutation Status.
Just a few years ago only the most sophisticated research hospitals are able to do the IgVH gene mutation status test. That is no longer the case. Several commercial testing labs now routinely do this test and the costs have come down significantly. IgVH status is probably the single most important prognostic test, along with FISH status. Not all local oncologists are familiar with the IgVH status test and you may have to do a bit of sweet-talking to get your guy to write a script for it. What you get in life depends to a great deal on your ability to negotiate with other people and getting effective medical care is no different. The good news is that IgVH gene mutation status does not change over time, it is cast in concrete for the rest of the patients CLL history. So, you do not have to get this test repeated (provided it was done right the first time!)
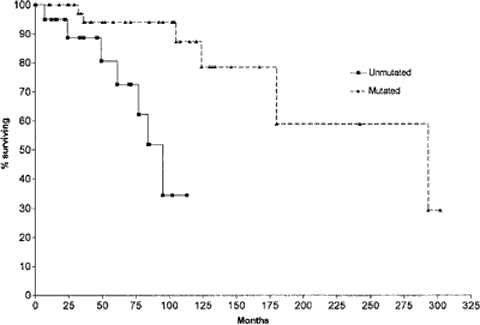
Unmutated IgVH Genes Are Associated With a More Aggressive Form of CLL
Source: Terry J. Hamblin, et al. Blood
Again, the title says it all: Unmutated IgVH Genes Are Associated With a More Aggressive Form of Chronic Lymphocytic Leukemia. The two curves compare Stage A CLL patients with mutated and unmutated VH genes. The difference in survival patterns could not be more dramatic.
Let me see if I can take some of the mystery out of IgVH gene mutation status. Remember, we said all cancers start with the first malignant cell, in our case the first malignant B-cell that is the great-grand-daddy of all the other CLL cells that came from it. Well, IgVH mutation status pinpoints the exact time in the life of this crucial first CLL cell when it became a malignant cell.
In some patients, this patriarch of the CLL clonal family became a malignant cell before it finished its "education" in the germinal center. This "ignorance" of the first CLL cell is reflected in the immunoglobin (Ig) of the cell being unchanged, it is just as virginal as it was when it was created in the bone marrow. Since this first cell eventually gave rise to all the other cells in the CLL clonal family, all of the CLL cells in these patients have unchanged or "unmutated" Ig. The mutation of interest is in the heavy chain of the Ig, in the variable region. Put all that together, and we come up with unmutated IgVH (immunoglobin, variable region, heavy chain).
In other patients, their very first CLL cell became a cancer cell after its trip to the germinal center. In these patients, since the first cell had graduated from the germinal center, the IgVH of this first CLL cell has been changed, (mutated), and this trait is carried forward by all the off-spring of this first cell. These patients have "mutated" IgVH.
In their extremely important study Hamblin, et al., studied 35 women and 49 men with classic CLL. 34 of the patients had progressive disease while 50 had stable disease. For each patient, the IgVH mutation status was determined. There are clear differences between the two groups of patients: those with unmutated IgVH had a more aggressive type of CLL than those with mutated IgVH. Unmutated IgVH was clearly associated with progressive disease and had a huge impact on survival statistics.
For example, comparing only stage A patients (see graph above), median survival was 95 months for the patients without mutations and 293 months for those with mutations. Just so you don't have to take out your calculator, 293 months is just under 25 years. Let's see now, the median age of the patients was around 63 years, add 25 years to that, and we get 88 years. CLL patients in this study with mutated IgVH gene lived on average to 88 years. For my money, I read these statistics to say that for this group of patients with mutated IgVH genes, CLL did not have any real impact on their overall life span. They probably lived just as long as their neighbors and friends that had never even heard of CLL.
But the story is different for the patients with unmutated IgVH. The median life expectancy for this group was 8 years. Since the age at diagnosis was the same for both groups, average patient in this group lived to be 63 + 8 = 71 years. That is a far cry from the 88 years of life for the IgVH mutated group.
Moral of the story, don't you wish every patient had CLL where the first nasty cancerous B-cell did not drop out of germinal school, bothered to finish its education and get its merit badge of IgVH gene mutation, before it decided to go bad and become a cancerous CLL cell?
Unfortunately for our three bucket division of patients, IgVH gene mutation status allows only a two bucket split. You either have the IgVH gene mutation (good news!!) or you don't (too bad, but it is not the end of the world).
CD38 is a marker that is expressed on CLL cells that are busy having babies and therefore their numbers are growing fast. There was a lot of excitement a couple of years ago when it was thought that the expression of the CD38 marker on CLL cells closely paralleled their IgVH gene mutation status. Unlike IgVH mutation status testing that requires a well equipped research lab, CD38 levels on CLL cells can be measured by straightforward flow cytometry.
Remember, we are interested in measuring the CD38 levels on just the CLL cells, not all cells. The measurement therefore requires that we first be sure we are talking of CLL cells only and then look for CD38 expression on these cells. Going back to the first section of this paper, we learned that co-expression of CD5 and CD19 is a classic pattern of CLL cells. In other words, if we focus only on cells carrying both the CD5 and the CD19 markers, and then look to see how many of them are carrying the CD38 marker as well, we have done the job right. In their pivotal paper Damle, et al., defined the cut-off point as 30%: if less than 30% of the CD5/CD19 positive cells (i.e., CLL cells) expressed CD38, that was good news. Patients whose CLL cells had more than 30% CD38 positive cells were likely to have poorer prognoses. As in the case of FISH and B2M, CD38 levels can and do change over time.
Closer examination of the data and additional work done by other labs and other researchers suggested that the correlation between IgVH gene mutation status and CD38 expression was not as good as one could have hoped. True, low CD38 expression on CLL cells was still an indicator of good prognosis and patients with high CD38 expression on their CLL cells typically had more aggressive disease and poorer survival odds. But the two indicators did not track each other exactly, and that was a disappointment. Also, the 30% cut-off level was questioned, several researchers felt a cut off of 20% or even 10% was a better choice.
Putting all this information together, we are not too far from reality if we use percentage of CD38 positive CLL cells as a semi-independent indicator. Patients with less than 20% CD38 positive CLL cells are in Bucket A. If they have between 20% and 30% CD38 they are in Bucket B; patients with more than 30% CD38 expression on CLL cells are in the ugly Bucket C.
This is the latest sexy new prognostic indicator to hit the wires a few years ago. So far, it looks set to become the indicator to measure, since it has a number of advantages:
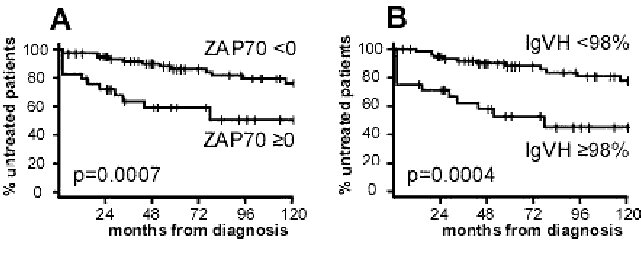
Impact of prognostic features on the clinical course of CLL
Source: ZAP-70 expression identifies a chronic lymphocytic leukemia subtype with unmutated immunoglobulin genes,
inferior clinical outcome and distinct gene expression profile Blood, February 20, 2026. Adrian Wiestner et al.
This chart shows the rate of disease progression as assessed by the treatment-free time interval measured in months from diagnosis.
Below is a table that summarizes all the details we have discussed above, and divides patients into the three Buckets. I have also included the classic prognostic indicator, Rai staging as well. You can get more detail of the Rai (and Binet) staging systems by going to: Staging Does Not Predict Survival.
Nothing in life is simple. For many of you not all of the prognostic indicators will line up exactly, making it crystal clear which bucket you belong to. You will have to use your judgment in how you deal with a split decision. But if you get all these prognostic testing done, I am willing to bet you have a much better idea of how your CLL is likely to progress.
| Prognostic Indicator | Bucket A | Bucket B | Bucket C |
| FISH | 13q deletion only | Normal karyotype or Trisomy 12 |
11q or 17p deletion |
| β2M | below 2.0 | 2.0 to 4.0 | above 4.0 |
IgVH |
Mutated | Mutated/Unmutated | Unmutated |
| CD38 | less than 20% |
between 20% and 30% | greater than 30% |
| ZAP-70 | less than 20% | about 20% | greater than 20% |
| ALC Doubling Time | longer than 1 year | about 1 year | shorter than 1 year |
| Rai Stage | Stage 0 or 1 | Stage 2 | Stage 3 or 4 |
| Action Plan |
|
|
|
I have said this before, but I want to repeat it so that there is no chance of you missing the point: Bucket C is not an automatic kiss of death. It just means you do not have the luxury of dithering, indulging in wishful thinking, or trying to cure an aggressive form of CLL with home remedies. Time you waste in exploring half measures and ill thought out strategies will be precious time that would have been better spent coming to pragmatic grips with the issues involved.
Similarly, if you are in lucky Bucket A, panicking at the mention of the word Cancer and rushing to over-treat your smoldering CLL are tantamount to spoiling your good fortune. If you are fortunate enough to have a smoldering variety of CLL, last thing you want is to screw up the lucky phenotype with aggressive chemotherapy. Many chemotherapy drugs are mutagenic and can hasten the process of clonal evolution, sufficient to kick you from your comfortable Bucket A home to more dangerous Buckets B or C.
If you are a control freak and just hate the concept of sitting on your hands waiting, may I suggest you use your energies to get your body into better shape? If you are a smoker, quit. If you are a couch potato, get into a pattern of regular and vigorous exercise. Learn to eat a healthy and balanced diet with plenty of fresh vegetables and fruits, cold water fish such as tuna. Stay out of the sun since CLL patients (even the good prognosis ones) are at increased risk of aggressive skin cancer. Make sure you cover all your other health bets (routine colonoscopy, mammogram, prostate test etc as recommended by your doctors). Infections are common in CLL patients, especially infections of the respiratory system. Uncontrolled pneumonia is the single biggest killer of CLL patients and you ignore pulmonary infections at your peril.
Bottom line, use the Wait time to get into fighting trim. Many of the drugs used to treat CLL are tough on the liver, kidneys, lungs and heart, all the organs of your body. You will survive cancer therapy so much better if you are otherwise in good health.
Last but not least, please do not write out your last will and testament based solely on the survival statistics presented here. For starters, these are historical statistics and we have every reason to believe things are improving for CLL patients with newer therapies and better understanding. Also, statistics are great for giving you the odds, but you may still be the one person that falls way outside the norms, you may be lucky and do a lot better (or a lot worse, if you are unlucky) than the statistics. No, it is not a crap shoot. You would not bet on a horse race without knowing the odds, would you? But you would not expect the odds to guarantee a win either. Same story here. Knowing the odds makes it easier for you to play the hand you have been dealt to best advantage. None of us can hope to do better than that.
 Enter Keywords: |
———
Disclaimer: The content of this website is intended for information only and is NOT meant to be medical advice. Please be sure to consult and follow the advice of your doctors on all medical matters.
Copyright Notice:
Copyright © 2026-2007 CLL Topics, Inc. All Rights Reserved.
All materials contained on this site are protected by United States copyright law and may not be reproduced, distributed, transmitted, displayed, published or broadcast without the prior written permission of CLL Topics, Inc. You may not alter or remove any trademark, copyright or other notice from copies of the content.
However, you may download and print material from CLLTopics.org exclusively for your personal, noncommercial use.
———


 The Bucket Brigade
The Bucket Brigade

