|
||||||||||
Date: June 6, 2026
by Chaya Venkat
Related Article:


Flavopiridol is an important experimental drug, derived from a medicinal plant from India which has been used for centuries in many indigenous medicines. Flavopiridol is being developed by Aventis Oncology in collaboration with the National Cancer Institute (NCI). Its use is presently under investigation for a variety of solid tumors as well as hematological cancers. Several very bright researchers whose horse sense I trust seem to think this drug has "legs", that it will survive the early phase optimization trials to become a major drug for the treatment of CLL.
To understand how flavopiridol works, you need to learn a little bit about the life cycle of cells. Just like human beings who go through birth, adolescence, reproductive stages, senescence and death, each and every cell in your body also goes through its own life cycle. As always, there is a huge amount of mind-bending detail that scientists are piecing together, but we can get the gist of it with a thumbnail sketch.
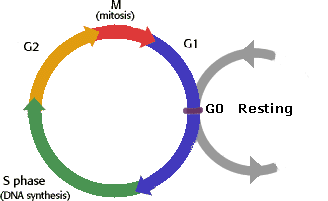
Here are the various stages of a cell's life. Let us start from the G0 stage. This is akin to childhood in our lives. G0 is the quiescent phase, the cell is immature and not ready to do much of anything. When it is ready to grow up and take part in real life, it has to pass a test to make sure everything is in order. This test is called a "checkpoint". In normal cells, if the cell does not pass the test, it is not allowed to progress to the next stage. This test prior to entry into G1 stage is called the G1 checkpoint. In many cells the G1 checkpoint seems to be the most important. If a cell passes G1, it will usually complete the cycle and divide. If it does not receive the go-ahead at G1, it will go back to the G0 resting phase. Most cells in adult human body are in the G0 phase.
Once the cell has met the standards of the G1 checkpoint, the cell grows in size, making more protein and synthesizing RNA. Think of this as the period of adolescence in humans, when the cell is getting ready for its adult role of reproduction. After completion of the G1 stage, the cell goes on to the S phase of its life, where "S" stands for synthesis. This is when the cell makes fresh DNA, making exact copies of each of its chromosomes. Think of this a the pregnancy phase, the cell is getting ready to give birth to its daughter. But before it is allowed to give birth, it has to go through another period called G2 phase, when it puts the finishing touches and pass/fail another test, the G2 checkpoint to determine if the cell is fit to reproduce. These three stages, G1, S and G2 are together called Interphase.
If the cell has met the stringent requirements of G1 and G2 checkpoints, it is allowed to proceed to Mitosis, akin to childbirth by our analogy. Cell growth and protein production stop, all other functions take a back seat as the cell focuses all of its energy on the complex job of dividing itself into two similar daughter cells. Mitosis is much shorter than Interphase, it lasts only about one to two hours. There is yet another checkpoint in the middle of mitosis that ensures the cell is ready to complete cell division, with no detectable screw-ups along the way. After completing the process of duplicating itself, the two daughter cells enters G0 resting stage, until they are ready once more to enter the cycle of G1, S, G2 and M. (You can find an animated graphical depiction of these cell cycle stages and the associated chromosomal activity at: http://www.cellsalive.com/cell_cycle.htm.)
As you can see, the rites of passage of cells from birth to the point where they have earned the right to replicate themselves is pretty strict, with multiple tests and checkpoints along the way. These are necessary hurdles, in order to make sure the genetic information transmitted to the next generation of daughter cells is correct and without errors. The checkpoints are important in weeding out potentially dangerous mutations that can give rise to cancer. With all these safeguards in place, how is it that humans do get cancer? Unfortunately, cancer cells are able to subvert the checks and quality controls and sneak past the checkpoints. For every system of checks and repeat quality control tests that our bodies can invent, there seem to be an equal number of ways of subverting the system. Cancer gets a foothold and establishes itself as a viable entity in our bodies only when it has learned a way of getting around the checkpoints of the cell cycle.
The kinases are proteins that control the cell cycle, but most of the time they are in an inactive form. To be active the kinases must be associated with another type of protein called cyclins. Hence they are called cyclin-dependent kinases or "Cdks". Cdks control the various checkpoints, they are the gatekeepers that decide whether to pass or fail cells as they go through the various quality control checkpoints in the cell cycle. Cyclins are frequently over-expressed in human cancers (for example, the tell-tale marker for Mantle Cell Leukemia is over expression of Cyclin D1), lowering the bar for the checkpoints, letting just any malignant cell to pass the checkpoint and on its way to reproduction. Another common feature of cancer cells is decreased in the function of inhibitors of Cdks. Think of these peptides (p16 and p21 are two important ones) as the supervisors that keep an eye on the Cdks, to make sure they do not drop the standards too low. When the supervisors are asleep on the job, and the Cdks get lax in their standards, malignant cells can pass the checkpoints and proliferate. This is one of the most common features of all cancers.
Initial preliminary investigations of flavopiridol in breast and lung cancer cell lines demonstrated that flavopiridol blocked cells from progressing beyond stages G1 and G2. It is as if the cells are frozen into endless childhood and adolescence, incapable of making the next generation of cells. Interestingly, flavopiridol has been shown to enhance the cytotoxic effect of conventional chemotherapeutic agents in gastric and breast cancer cell lines. To bring it close to home, patients with deletions or defects in the ATM and/or TP53 genes (11q or 17p deletions: Cytogenetics of ATM and P53) have multiple defects in both G1 and G2 checkpoint controls. This is why the ATM and TP53 genes are considered the keepers of the genome, defects in these genes allow uncontrolled reproduction of cancer cells.
While it is clear that flavopiridol blocks cells from subverting the cell cycle checkpoints, and therefore from proliferating, it is not entirely clear how it actually kills cells. Some researchers believe that flavopiridol suppresses expression of bcl-2, a cancer promoter that is responsible for making cancer cells hard to kill. Another area of ongoing research is establishing safe method for administering this powerful drug, as well as the right dosage. Toxicity is of concern, as well as tumor lysis syndrome. Below we will review an ASCO abstract (I am reviewing this abstract before its publication in this year's ASCO meeting!) as well as a pivotal paper by Drs. Byrd, Grever et al, since both are particularly relevant to the use of flavopiridol in CLL. Links and abstracts of several other articles of interest are given at the end of this review for the interested reader.
The abstract of the Byrd, Grever, et. al. paper is given below, as well as the link where you can read the full text of the article, if you are so inclined. This is a timely review of the technology. There are hints that the CALGB (Cancer and Leukemia Group B - www.calgb.org) may soon initiate clinical trials of flavopiridol as a way of clearing out last traces of CLL. Given recent concerns regarding use of Campath for similar MRD (Minimum Residual Disease) consolidation therapy (Hitting a Home Run with Campath Consolidation?), this is an important development. Of particular importance to poor prognostic group CLL patients is the clear indication that flavopiridol does not seem to need proper functioning of ATM or TP53 genes in order to do its job, a valuable and rather rare feature for our Bucket C folks.
Blood Journal Article (Free Full-text)
Blood. 1998 Nov 15;92(10):3804-16.
Flavopiridol induces apoptosis in chronic lymphocytic leukemia cells via activation of caspase-3 without evidence of bcl-2 modulation or dependence on functional p53.
Byrd JC, Shinn C, Waselenko JK, Fuchs EJ, Lehman TA, Nguyen PL, Flinn IW, Diehl LF, Sausville E, Grever MR.
Division of Hematology-Oncology, Walter Reed Army Medical Center, Washington, DC.
Flavopiridol has been reported to induce apoptosis in lymphoid cell lines via down regulation of bcl-2. The in vitro activity of flavopiridol against human chronic lymphocytic leukemia (CLL) cells and potential mechanisms of action for inducing cytotoxicity were studied. The in vitro viability of mononuclear cells from CLL patients (n = 11) was reduced by 50% at 4 hours, 24 hours, and 4 days at a flavopiridol concentration of 1.15 micromol/L (95% confidence interval [CI] +/-0.31), 0.18 micromol/L (95% CI +/-0.04), and 0.16 micromol/L (95% CI +/-0.04), respectively. Loss of viability in human CLL cells correlated with early induction of apoptosis. Exposure of CLL cells to 0.18 micromol/L of flavopiridol resulted in both decreased expression of p53 protein and cleavage of the caspase-3 zymogen 32-kD protein with the appearance of its 20-kD subunit. Contrasting observations of others in tumor cell lines, flavopiridol cytotoxicity in CLL cells did not correlate with changes in bcl-2 protein expression alterations. We evaluated flavopiridol's dependence on intact p53 by exposing splenocytes from wild-type (p53(+/+)) and p53 null (p53(-/-)) mice that demonstrated no preferential cytotoxicity as compared with a marked differential with F-ara-a and radiation. Incubation of CLL cells with antiapoptotic cytokine interleukin-4 (IL-4) did not alter the LC50 of flavopiridol, as compared with a marked elevation noted with F-ara-a in the majority of patients tested. These data demonstrate that flavopiridol has significant in vitro activity against human CLL cells through activation of caspase-3, which appears to occur independently of bcl-2 modulation, the presence of IL-4, or p53 status. Such findings strongly support the early introduction of flavopiridol into clinical trials for patients with B-CLL.
PMID: 9808574
___________
The authors point out that one of the major hurdles of treating CLL is that most patients (except the lucky long-term smolderer) do "evolve", gradually adding more and more complex and dangerous chromosomal aberrations. As the disease progresses, it gradually acquires resistance to most of the common chemotherapy drugs available. Defects in TP53 gene (and to some extent its side-kick the ATM gene) means "virtually no responses to either alkylator or purine analog therapy". Indeed, some of these dangerous mutations may arise as a consequence of prior therapy (Fludarabine Monotherapy Is No Longer the Gold Standard). This paper demonstrates marked cytotoxicity of flavopiridol toward human CLL cells, circumventing drug resistance incurred by a p53 mutation. Because drug resistance is more frequently observed in previously treated patients, it is good to see that "in vitro sensitivity to flavopiridol is not altered by prior treatment and more advanced disease". Here are the highlights of this preclinical work. Remember, this pre-clinical work is with cells in the laboratory, not in live human patients.
ASCO abstract.
Flavopiridol
Moran, D Lucas, K Cunningham, A Colevas, MR Grever, JC Byrd.
The Ohio State University, Columbus, OH and National Cancer Institute, Washington DC.
CLL patients (pts) with del(17p13) [region corresponding to the p53 gene] have an extremely poor outcome and are not responsive to most conventional therapies (fludarabine, chlorambucil, and rituximab). Pre-clinical studies demonstrated flavopiridol induces apoptosis in CLL cells in a p53-independent manner when incubated in media utilizing fetal calf serum (FCS). Subsequent phase I/II studies using a 24-72-hour continuous IV schedule with flavopiridol failed to demonstrate activity in CLL and other cancers. Discordant binding of flavopiridol to human plasma proteins as compared to FCS prompted pharmacokinetic modeling from the continuous IV infusion studies demonstrating a dosing schedule of 30-minute IV bolus followed by 4-hour continuous infusion would be optimal. We report preliminary results of an ongoing phase I dose escalation study of single agent flavopiridol utilizing this schedule. Nine patients (median age 60, range 44-73) with fludarabine-refractory CLL have been enrolled (median prior number of therapies 7, range 2-13). Five patients had del(17p), three pts had bulky Rai stage I disease, and 6 pts were stage III/IV at time of study entry. Pts received 50% of the flavopiridol dose by 30-minute IV bolus, followed by the remaining 50% given by 4-hour infusion, weekly for 4 doses, on a 6-week cycle. Six pts in cohort 1 received 60 mg/m2/dose, and 3 pts in cohort 2 received 80 mg/m2/dose. Six pts developed transient grade 4 neutropenia, and grade 3-4 toxicity was also observed for thrombocytopenia (1), dyspnea (1), edema (1) and diarrhea (1). Other toxicities were common but grade 1-2 only (fatigue 7, nausea 6, anorexia 6). In cohort 2, three highly refractory patients developed life threatening tumor lysis syndrome. Two pts developed tumor lysis to the first dose of flavopiridol. One pt was able to complete his first cycle without further tumor lysis. The second of these 2 patients died of overwhelming tumor lysis to her first dose, with predominant hyperkalemia not responding to forced alkaline hydration and IV insulin, and subsequent cardiac arrhythmia and asystole. A third pt developed tumor lysis to the first dose of cycle 2, which responded to medical management. This pt also developed hypotension that did not respond to IV fluids but resolved with steroid therapy, suggesting a possible cytokine release syndrome. Pts completed a median of 1 cycle (range 0-2). The study was placed on hold, in order to develop and institute guidelines to prevent further episodes of life threatening tumor lysis. Treatment was stopped due to disease progression (3), infection (1), tumor lysis (2), and temporary discontinuation of study (3). Two pts in cohort 1 with fludarabine-refractory CLL experienced an NCI 96 partial response without significant toxicity that has persisted for 5+ months. One of these pts has a del(17p13). In summary, single agent flavopiridol given weekly by 30-minute IV bolus followed by 4-hour infusion in pts with highly refractory CLL, has significant clinical activity, as evidenced by responses in fludarabine-refractory CLL and acute tumor lysis syndrome. Further study of flavopiridol in CLL and other B-cell diseases utilizing this pharmacokinetically modeled schedule is warranted.
The above ASCO abstract (courtesy of Dr. John Byrd, MD) makes for somewhat disappointing reading, and I must also admit to a level of alarm. As expected, there was significant toxicity, and tumor lysis syndrome observed in this human clinical trial. But I expected far greater response as quid-pro-quo for these deficits. To get a better handle on these results as well as get a heads-up on future plans for flavopiridol, I contacted Dr. John Byrd with my usual laundry list of questions. His response was both prompt and courteous. The bullet points below are based on my understanding of our email exchange, not direct quotes attributable to Dr. Byrd. Any errors or omissions are therefore entirely attributable to me.
I gather that:
There is little doubt that flavopiridol is an extremely potent drug, and of particular interest to CLL patients in late stage disease or with poor prognosis cytogenetics. Not only does it seem to inhibit proliferation by preventing subversion of the checkpoint system of the cell cycle, it is also able to deliver the death blow, cause actual cell kill. The single biggest advantage of this drug seems to be its ability to do this without depending on the ATM and/or TP53 genes. Since most late stage and/or poor prognosis CLL patients have ATM / TP53 defects, and are therefore also resistant to standard chemotherapy drugs, flavopiridol is an important addition to our arsenal.
Of significant concern is the apparent lack of selectivity, since flavopiridol seems to kill cells from healthy volunteers just as efficiently as it does cells from CLL patients. This non-selective cell kill may be responsible for therapy associated toxicity and side effects seen in the human trial reported in the ASCO abstract above. Also of concern to this layperson reporter is the extreme rapidity with which cells are killed after exposure to flavopiridol. This suggests that cell kill may be so rapid that it may out strip the body's ability to deal with the consequences of massive cell kill and cytokines released as a consequence, and this seems to have been verified in the significant number of patients who developed tumor lysis syndrome. Dosage and method of administration are being fine-tuned, as well as appropriate prophylactic medications to protect against toxicity and immune suppression. Concerns about tumor lysis suggests that the drug should probably be used in the setting of low level tumor burden, as in MRD situations or very early stage disease in patients with poor prognosis cytogenetics. I am happy to see the researchers are indeed incorporating MRD setting into their next clinical trial.
In the final analysis, it is always a tradeoff between administering enough of the drug to get significant activity against the tumor, but not so much that there are unacceptable level of toxicity and related problems. I would like to see incorporated into the new clinical trial design adjuvants that work synergistically with flavopiridol, such that the dosage can be reduced further, improving the bang for the buck of this potent drug. After all, it is no fun to cure the CLL but the patient ends up dead any way.
Please keep in mind that there is always a learning curve in using these powerful chemotherapy drugs: as we learn more about dosage, timing, prophylaxis and administration methods, the results typically become more acceptable. Further, there is a whole world of synergy and unexpected interactions that need to be explored before we can say we know how to use a given agent.
Pre-clinical data from the Byrd, Grever, et. al. paper reviewed above suggest that there is reason to expect synergy between flavopiridol and some of the conventional chemotherapy drugs. However, patients who volunteer for the soon-to-be announced clinical trial would already have had RPC (or one of the other chemo-immunotherapy combo regimens) thrown at them, followed by the flavopiridol for MRD clean-up. How many more chemotherapy drugs and drug-induced toxicity can these patients absorb? I would like to challenge the researchers to consider a less scary alternative, adjuvants that have synergy with flavopiridol but do not necessarily add to the toxicity bill.
Here is an example, I am sure the experts can come up with many more and perhaps better scenarios. It so happens that one of the major mechanisms of resistance to flavopiridol is telomerase activation (see the abstract below). If you do not know what telomerase activation is, do not worry. That is the fascinating subject of my next article, so just hold your horses for a while. For now, all you need to know is that telomerase activation is what makes cancer cells almost immortal, lets them make too many babies, makes them hard to kill, and a royal pain in all sorts of other ways. Telomerase activation is also used by several viruses in order to protect their homes in infected cells, grow their neighborhoods by creating even more infected cells. Two famous examples are the AIDS virus which infects T-cells, and the EBV (Epstein-Barr virus) involved in mononucleosis, where B-cells are infected. The plot thickens further, since it has just been shown by Damle, Chiorazzi, et. al. that telomerase activation is most common in poor-prognosis CLL patients, those with unmutated IgVH genes (second abstract below).
Mol Pharmacol. 2026 Nov;64(5):1101-8.
Acquired cellular resistance to flavopiridol in a human colon carcinoma cell line involves up-regulation of the telomerase catalytic subunit and telomere elongation. Sensitivity of resistant cells to combination treatment with a telomerase inhibitor.
Incles CM, Schultes CM, Kelland LR, Neidle S.
The School of Pharmacy, 29-39 Brunswick Square, London WC1N 1AX, UK.
Flavopiridol is a broad-spectrum inhibitor of cyclin-dependent kinases and of global transcription via the inhibition of positive transcription elongation factor b (P-TEFb). Although flavopiridol is currently undergoing phase II clinical trials, acquired cellular resistance to the compound during treatment is a potential problem, as it is with almost all current anticancer agents. A HCT116 human colon carcinoma cell line with an acquired 8-fold resistance to flavopiridol has been established. We report here that there are changes in these resistant cells in terms of telomere length and telomerase activity, whereas no change in the expression of the P-TEFb subunits CDK9, cyclin T1, cyclin T2a, or cyclin T2b was observed. The level of mRNA expression for the telomerase catalytic subunit hTERT was increased over 2-fold in the resistant cells, and mean telomere length was found to be 2 kb longer than the parental length, although telomerase activity was unchanged. The level of mRNA expression for the telomeric binding protein Pot1 was also increased. We also report that treatment of HCT116 cells with a combination of the G-quadruplex interacting telomerase inhibitor BRACO-19 and flavopiridol results in a 3-fold decrease in population doubling and prevents recovery from treatment with either compound alone. Treatment of flavopiridol-resistant cells with BRACO-19 alone also led to rapid inhibition of cell growth, which is not observed in the parental line. The finding that only the resistant line, with up-regulated telomerase, responds to this G-quadruplex inhibitor is consistent with the hypothesis that the mechanism of BRACO-19 down-regulation of cell growth directly involves the targeting of telomeres and telomerase.
PMID: 14573759
____________
Blood. 2026 Jan 15;103(2):375-82. Epub 2026 Sep 22.
Telomere length and telomerase activity delineate distinctive replicative features of the B-CLL subgroups defined by immunoglobulin V gene mutations.
Damle RN, Batliwalla FM, Ghiotto F, Valetto A, Albesiano E, Sison C, Allen SL, Kolitz J, Vinciguerra VP, Kudalkar P, Wasil T, Rai KR, Ferrarini M, Gregersen PK, Chiorazzi N.
North Shore-Long Island Jewish Research Institute, 350 Community Dr, Manhasset, NY.
Patients with B-cell chronic lymphocytic leukemia (B-CLL) segregate into subgroups with very different survival times. Because clinical observations suggest that leukemic cells accumulate at different rates, we measured telomere length and telomerase activity in B-CLL cells to distinguish differences in cellular replication. Our data indicate that the telomeres of B-CLL cells are shorter than telomeres of B cells from healthy subjects, indicating that the leukemic cells have a prolonged proliferative history. Leukemic cells of the immunoglobulin V gene mutation subgroups differ in telomere length and telomerase activity. B lymphocytes from the subgroup with poor outcome and with limited IgV gene mutations have uniformly shorter telomeres and more telomerase activity than those from the subgroup with better outcome and with considerable mutations. Differences in telomere length appear to largely reflect the proliferative histories of precursors of the leukemic cells, although differences in cell division, masked by the action of telomerase, cannot be excluded. These results may provide insight into the stages of maturation and the activation pathways of the cells that give rise to B-CLL. In addition, they reinforce the concept that B-CLL is not simply an accumulative disease of slowly dividing B lymphocytes but possibly one of B cells with extensive proliferative histories.
PMID: 14504108
____________
Several candidate drugs for telomerase inhibition have been put through their paces in AIDS research, with clinical trials conducted and data available for review. Since telomerase activation seems to be one of the major weaknesses of flavopiridol, would it make sense to combine flavopiridol with one of the telomerase inhibitors? Would the synergy between the telomerase inhibitor and flavopiridol mean that a lower dosage of the flavopiridol can be used to get the same response? What are good telomerase inhibitors? I am glad you asked. A famous telomerase inhibitor you might have heard about is AZT, a drug used in AIDS therapy. In addition, you would be pleased to know that EGCG, one of the active components in green tea and a favorite topic of discussion on CLL Topics, is also an effective telomerase inhibitor.
Hmmm. Does this suggest including green tea EGCG as an adjuvant in the flavopiridol regimen may prove synergistic? Flavopiridol is derived from an Indian medicinal plant, and green tea is from Japan and China. Folks, looks like we are talking of an Asian trend here. You can read the full article describing the connection between EGCG and telomerase inhibition by clicking on the link provided below.
Article from Cancer Research Journal (Free full-text)
Cancer Res. 2026 Feb 15;63(4):824-30.
Blocking telomerase by dietary polyphenols is a major mechanism for limiting the growth of human cancer cells in vitro and in vivo.
Naasani I, Oh-Hashi F, Oh-Hara T, Feng WY, Johnston J, Chan K, Tsuruo T.
Cancer Chemotherapy Center, Japanese Foundation for Cancer Research, Kami-Ikebukuro, Toshima-ku, Tokyo 170-8455, Japan.
Animal and epidemiological studies reveal that consuming food and beverages rich in polyphenols (e.g., catechins, flavones, and antocyanines) is associated with a lower incidence of cancer, and several molecular mechanisms have been proposed for explaining this effect. However, because most of these mechanisms were observed only under specific and nonphysiological conditions, and in most cases, with practically irrelevant concentrations, there is still no clear-cut or universal explanation for the major events that underlie the anticancer effects of polyphenols. In this study we present clear in vitro and in vivo evidence that the inhibition of the cancer-associated enzyme telomerase is a key mechanism involved in cancer inhibition by epigallocatechin gallate (EGCG), a major tea polyphenol. We demonstrate that EGCG and other selected polyphenols undergo structural rearrangements at physiologically permissible conditions that result in remarkably increased telomerase inhibition. In nude mice models bearing both telomerase-dependent and -independent xenograft tumors cloned from a single human cancer progeny, only the telomerase-dependent tumors responded to prolonged oral administration of EGCG. Thus, EGCG and likely other structurally related dietary polyphenols seem to act as prodrug-like molecules that, once ingested and distributed, undergo structural changes that favor potent activity against telomerase.
PMID: 12591733
____________
Article from Blood Journal (Free full-text)
Blood. 1997 Dec 1;90(11):4307-12.
The novel cyclin-dependent kinase inhibitor flavopiridol downregulates Bcl-2 and induces growth arrest and apoptosis in chronic B-cell leukemia lines.
Konig A, Schwartz GK, Mohammad RM, Al-Katib A, Gabrilove JL.
The Molecular Therapeutics Program, the Sloan-Kettering Institute, New York, NY.
Flavopiridol is a novel, potent inhibitor of cyclin-dependent kinases (CDK). This synthetic flavone has been reported to exhibit antitumor activity in murine and human tumor cell lines in vitro and in vivo and is currently undergoing clinical phase I evaluation. In the present study, 1 Epstein-Barr virus (EBV)-transformed B-prolymphocytic cell line (JVM-2), 1 EBV-transformed B-CLL cell line (I83CLL), and 1 non-EBV transformed B-CLL cell line (WSU-CLL) were used as targets. Treatment of the cells with flavopiridol (100 nmol/L to 400 nmol/L) led to a marked dose- and time-dependent inhibition of cell growth and survival as determined using trypan blue exclusion. Morphologic analysis showed characteristic apoptotic changes such as chromatin condensation and fragmentation, membrane blebbing, and formation of apoptotic bodies. Furthermore, quantitative assessment of apoptosis-associated DNA strand breaks by in situ TdT labeling showed that a significant number of flavopiridol-treated cells underwent apoptosis. These cellular effects were associated with a significant decrease in bcl-2 expression as observed by Northern and Western blotting. The results showed that flavopiridol downregulates bcl-2 mRNA and bcl-2 protein expression within 24 hours. Genistein and quercetin, two flavonoids that do not inhibit CDKs, did not affect bcl-2 expression. These data suggest an additional mechanism of action of this new flavone which might be useful as an agent in the treatment of chronic lymphoid malignancies.
PMID: 9373241
___________
Article from Cancer Spectrum the Journal of the NCI (Free full-text)
J Natl Cancer Inst. 2026 Mar 1;92(5):376-87.
Preclinical and clinical development of cyclin-dependent kinase modulators.
Senderowicz AM, Sausville EA.
DTP Clinical Trials Unit, Developmental Therapeutics Program, Division of Cancer Treatment and Diagnosis, National Cancer Institute, Bethesda, MD.
In the last decade, the discovery and cloning of the cyclin-dependent kinases (cdks), key regulators of cell cycle progression, have led to the identification of novel modulators of cdk activity. Initial experimental results demonstrated that these cdk modulators are able to block cell cycle progression, induce apoptotic cell death, promote differentiation, inhibit angiogenesis, and modulate transcription. Alteration of cdk activity may occur indirectly by affecting upstream pathways that regulate cdk activity or directly by targeting the cdk holoenzyme. Two direct cdk modulators, flavopiridol and UCN-01, are showing promising results in early clinical trials, in which the drugs reach plasma concentrations that can alter cdk activity in vitro. Although modulation of cdk activity is a well-grounded concept and new cdk modulators are being assessed for clinical testing, important scientific questions remain to be addressed. These questions include whether one or more cdks should be inhibited, how cdk inhibitors should be combined with other chemotherapy agents, and which cdk substrates should be used to assess the biologic effects of these drugs in patients. Thus, modulation of cdk activity is an attractive target for cancer chemotherapy, and several agents that modulate cdk activity are in or are approaching entry into clinical trials.
PMID: 10699068
____________
Leuk Lymphoma. 2026 Feb;44(2):337-42.
Flavopiridol induces apoptosis in B-cell chronic lymphocytic leukaemia cells through a p38 and ERK MAP kinase-dependent mechanism.
Pepper C, Thomas A, Fegan C, Hoy T, Bentley P.
Department of Haematology, Llandough Hospital, Penlan Road, Penarth, Vale of Glamorgan, CF64 2XX, UK.
Flavopiridol, a synthetic flavone, has been previously shown to induce apoptosis in B-cell chronic lymphocytic leukaemia (B-CLL) cells in vitro. The apoptosis was associated with a concomitant activation of caspase-3 without evidence of dependence on functional p53 or Bcl-2 family modulation. In this study, we examined flavopiridol-induced apoptosis in terms of upstream caspase activity, cell cycle distribution and signal transduction, in order to elucidate the mechanism of action of this potent cytotoxic agent. Flavopiridol-induced apoptosis was significantly abrogated by the caspase-9 inhibitor Z-LEHD-FMK (p = 0.002; paired t-test) but was not altered by the caspase-8 inhibitor Z-IETD-FMK (p = 0.37; paired t-test). There was a concentration-dependent increase in a sub G0/G1 peak indicative of apoptotic cells but if these cells were excluded by gating no other cell cycle perturbations were observed suggesting that flavopiridol is capable of inducing apoptosis in cells in all phases of the cell cycle. Significantly, apoptosis was associated with activation of p38 MAP kinase and suppression of ERK activity (p = 0.0036 and p = 0.0048, respectively; paired t-test). These results show for the first time that flavopiridol modulates specific cellular signal transduction pathways in B-CLL cells thereby altering the balance between survival and cell death signals and providing a rationale for the p53-independent nature of flavopiridol-induced apoptosis. Further work is required to identify whether combinations of conventional chemotherapeutic drugs and novel agents like flavopiridol can be used to improve patient outcomes in the treatment of B-CLL.
PMID: 12688354
____________
Article from 2026 ASH Education Book (Free full-text)
Hematology (Am Soc Hematol Educ Program). 2026;:443-62.
Rational approaches to design of therapeutics targeting molecular markers.
Klasa RJ, List AF, Cheson BD.
Division of Medical Oncology, British Columbia Cancer Agency, Vancouver, BC, Canada.
This paper introduces novel therapeutic strategies focusing on a molecular marker relevant to a particular hematologic malignancy. Four different approaches targeting specific molecules in unique pathways will be presented. The common theme will be rational target selection in a strategy that has reached the early phase of human clinical trial in one malignancy, but with a much broader potential applicability to the technology. In Section I Dr. Richard Klasa presents preclinical data on the use of antisense oligonucleotides directed at the bcl-2 gene message to specifically downregulate Bcl-2 protein expression in non- odgkin's lymphomas and render the cells more susceptible to the induction of apoptosis. In Section II Dr. Alan List reviews the targeting of vascular endothelial growth factor (VEGF) and its receptor in anti-angiogenesis strategies for acute myeloid leukemia (AML) and myelodysplastic syndromes (MDS). In Section III Dr. Bruce Cheson describes recent progress in inhibiting cell cycle progression by selectively disrupting cyclin D1 with structurally unique compounds such as flavopiridol in mantle cell lymphoma as well as describing a new class of agents that affect proteasome degradation pathways.
PMID: 11722998
____________
Article from Blood Journal (Free full-text)
Blood. 2026 Jul 15;96(2):393-7.
Protein kinase inhibitors flavopiridol and 7-hydroxy-staurosporine down-regulate antiapoptosis proteins in B-cell chronic lymphocytic leukemia.
Kitada S, Zapata JM, Andreeff M, Reed JC.
Burnham Institute, Program on Apoptosis and Cell Death Research, La Jolla, CA.
Compounds that inhibit protein kinases are currently undergoing clinical evaluation for the treatment of a variety of malignancies. The kinase inhibitors flavopiridol and 7 hydroxy-staurosporine (UCN-01) were examined for their effects on B-cell chronic lymphocytic leukemia (B-CLL) cells in vitro (n = 49). Flavopiridol and UCN-01 induced concentration-dependent apoptosis of most B-CLL samples tested, with greater than 50% cell killing occurring at concentrations of less than 1 mcmol/L, and with flavopiridol displaying more potent activity than UCN-01. Flavopiridol (0.1 mcmol/L) and UCN-01 (1 mcmol/L) also induced striking decreases in the levels of the antiapoptosis proteins Mcl-1, X-linked inhibitor of apoptosis (XIAP), and BAG-1 in nearly all cases of B-CLL and of Bcl-2 in approximately half of B-CLL specimens evaluated. In contrast, expression of the proapoptotic proteins Bax and Bak was not significantly influenced by these kinase inhibitors. Flavopiridol-induced decreases in the levels of antiapoptosis proteins Mcl-1 and XIAP preceded apoptosis and were not substantially affected by the addition of caspase inhibitors to cultures. In contrast, UCN-01-stimulated decreases in antiapoptosis proteins were slower, occurred concurrently with apoptosis, and were partially prevented by caspase inhibitors. The findings suggest that flavopiridol and UCN- 1 induce apoptosis of B-CLL cells through different mechanisms. The potent apoptotic activities of flavopiridol and UCN-01 against cultured B-CLL cells suggest that they may be effective as single agents in the treatment of B-CLL or for sensitizing B- LL cells to conventional cytotoxic drugs.
PMID: 10887097
_____________
You can also read about how flavopiridol affects the cell cycle in a review article on flavopiridol originally published in 1999 in Cancer Control, the journal of the Moffitt Cancer Center available through Medscape Today. (You will remember that registration in Medscape is free — and we highly recommend it. We do have an open registration for our members if you have a problem: User ID: CLLTopics ; password: optimist. But do yourself a favor and get your own registration. It is a useful entry to a valuable resource and that way Medscape will get a better count of its visitors.)
 Enter Keywords: |
———
Disclaimer: The content of this website is intended for information only and is NOT meant to be medical advice. Please be sure to consult and follow the advice of your doctors on all medical matters.
Copyright Notice:
Copyright © 2026-2007 CLL Topics, Inc. All Rights Reserved.
All materials contained on this site are protected by United States copyright law and may not be reproduced, distributed, transmitted, displayed, published or broadcast without the prior written permission of CLL Topics, Inc. You may not alter or remove any trademark, copyright or other notice from copies of the content.
However, you may download and print material from CLLTopics.org exclusively for your personal, noncommercial use.
———