|
|
|
|
Chemotherapy
Exciting New Drugs on the CLL HorizonSeptember 28, 2026 by Chaya Venkat The Light at the End of the Tunnel Is Getting BrighterSeveral recent research articles have opened up exciting new opportunities in the treatment of CLL. These feature new small molecule drugs that take an entirely different approach to targeting CLL cells. These are not your father's chemotherapy drugs, most of which target the nuclear DNA of cells, with the unavoidable risk of mutagenicity. Nor are they bulky (and expensive!) monoclonal antibodies like Rituxan and Campath, which have a hard time dealing with bulky lymph nodes and bone marrow - to some degree because it is hard for these "fat ladies" to get into all the nooks and crannies of bone marrow and lymph nodes. Small molecules like the ones we discuss below are more likely to reach all these hard-to-reach parts. Here is the best part: in this new approach to treating CLL, it makes no difference if you are IgVH mutated or unmutated, CD38 positive or not, chemo-naïve or have been through the wars with every chemotherapy drug known to man. In fact, there are some indications that heavily pretreated and late Rai stage patients may respond better to this approach! Stay with me on this story folks. These drugs have really caught the attention of some terrific researchers and they are on a fast track at a number of top notch research establishments (Dana Farber, M. D. Anderson and the Mayo Clinic, to name a few). My bet is that you will see at least a few of these in action in 2026 in several clinical trials. Some of these drugs are well known for other uses, some are quite old and even off-patent. Which brings us to the tricky question, who is going to push for their development? Money makes the world go around, we are all sufficiently grown up to know that. My worry is that without the backing of deep-pockets pharmaceutical companies with a vested interest, some of these drugs may die for lack of backing and never make it from the lab to the patient's bed side. That is where we come in, if we are smart about it and can mobilize as a group. After all we have more "skin" in this game than anyone else, right? First I have to give you a thumbnail sketch of the science. Not to worry, it is not that hard to understand and I will leave out all the details that you don't have to know, sort of give you the cartoon version. A Long, Long Time Ago..The story starts a long, long time ago, say a few billion years ago, give or take. Life consisted of very primitive single cells, barely managing to make a go of it. Then, some enterprising cell made a pact of cooperation with another tiny piece of life called a mitochondrion (plural, mitochondria). The deal was this. The mitochondria could live inside the cell. In addition to rent-free accommodations, the cell would also provide food, air, garbage removal and help when the mitochondria decides to have babies. The ultimate bed & breakfast, and lunch and dinner and ob-gyn help. In return, the mitochondria would have to do the one thing it had figured out how to do very well, convert food to energy, make sure the cell has enough energy to do all its household chores, as well as be fed well enough to have babies itself when it wants to. Thus began the most successful partnership ever struck on this earth. And since that first cell which made this deal was a good mother, every time it had a baby it made sure the baby also had its own resident baby mitochondria. Since cells without mitochondria got only about 4 units of energy per glucose molecule, and those with resident mitochondria had as much as 30-32 units of energy for the same amount of glucose, you can see why the newfangled idea caught on like wildfire, and the cells that could not adapt to the new technology died out. To this day, all multi-cellular organisms like you and me have mitochondria in each and every cell, not much changed over the billions of years. In fact, each cell may have many mitochondria. These little "power plants" in each cell are passed on from mother to daughter, your mother gave a copy of hers to you, in each and every cell of your baby body. Fathers have little to do with it, 99.99% of the blueprints for mitochondria come from the mother. (Sorry, guys. You got sort of side-lined on that one by Mother Nature). That means most humans (and giraffes and sea snails and polar bears and petunias) have very similar mitochondria, not enough difference worth discussing. That is one reason why we do not have to worry about matching mitochondrial DNA when we talk about blood transfusions or bone marrow / organ transplants, since we all have pretty near identical mitochondrial DNA. Cellular DNA is a different matter, since we do mix it up a great deal every time a new baby is conceived, half the DNA comes from the father and half from the mother, allowing for many variations on the theme. Isn't sex wonderful? Think of a cartoon picture of a B-cell. Inside the cell is a nucleus, which is where the all important cellular DNA resides, the mastermind that controls everything. This cellular DNA is a hugely complex affair, it has to take care of all the functions of the cell. Since it is so complex and damage to it is so catastrophic, we have developed very powerful and complex mechanisms for protecting it from all sorts of damage, radiation, chemotherapy and so on (see ATM and P53, guardians of the genome). Obviously, cancer cells also become very good at using these defense mechanisms to their own advantage, which is why they are so hard to kill. No question about it, the nucleus of the B-cell is like a strong and well guarded fortress, and the strands of cellular DNA are the crown jewels, hard to take down. Mitochondria, on the other hand, have lived a sheltered life these many billions of years. Nicely cocooned inside the cell, they have their own "digs" defined by a couple of sturdy membranes. Never having to lift a finger to make a living or find their own food, and not having to do much except the one major task of providing power for their host cell, they have had little reason to build any defenses. The cell provides glucose and oxygen, the mitochondria put out energy and water. That's the deal. Mitochondrial DNA is therefore a simple affair, short and sweet, with none of the complex safety features and defenses that you are likely to find in the cellular DNA. This is only a cartoon version of how mitochondria and cells cooperate. If you wish to learn more, here is good link with nice pictures and stuff: http://www.cytochemistry.net/Cell-biology/mitoch1.htm. How Does This Help Define a Cancer Therapy Strategy?By now you must have guessed where I am going with this. Taking down the cellular DNA tucked away in the fortress (nucleus) is a tough thing to do. But what about the poorly guarded power plants that are within the cell? What if we can sabotage the mitochondria and blow up the power plants? For starters, the cell will die for the simple reason it has no more power, it will starve to death. Another reason is that power plants are notorious for producing and storing all sorts of toxic waste. Dump all that toxic garbage into the cell and the cell is quickly poisoned beyond redemption. It does not matter what the mastermind cellular DNA has to say about it, mitochondrial death is hard to reverse. When its mitochondria die, the cell dies with them. That, in a nutshell, is the new (actually not so new, the concept has been around for a while) approach to treating CLL and presumably other cancers. Think like a terrorist - don't go after the hard-to-get cellular DNA living in the nucleus, go after the 'softer' target of mitochondria. Here are some approaches that are showing promise, all of them deal with the common theme of mitochondrial damage. 1. Increase the Garbage Production Mitochondria are normally very efficient at converting glucose and oxygen into power the cell can use, the major by-product being water; this is very efficient usage of oxygen, but not quite perfect. One of the waste products they produce is extremely reactive oxygen species such as peroxides. ROS ("Reactive Oxygen Species") are toxic because they can attack just about any thing. As the mitochondria get old and worn out, their efficiency decreases, possibly because of damage by their own ROS, and they get into a vicious cycle of making more and more of ROS. Cancer cells are inherently wasteful and spendthrift with energy, they are selfish and not good team players looking out for everyone's welfare. Their energy requirements go up tremendously especially when they are proliferating. Mitochondria in cancer cells are therefore often driven to ever-increasing loads, which means ever-increasing amounts of ROS, with ever-increasing damage to the mitochondrial DNA. Chemotherapy drugs also do a number on the mitochondria. Remember, we said cellular DNA is well protected, but not so mitochondrial DNA. So it is possible for some cancer cell to survive chemotherapy, but at the expense of damage to their mitochondrial DNA. In an elegant piece of research, researchers at M. D. Anderson have connected the dots and shown that CLL patients who have undergone chemotherapy are almost sure to have damaged mitochondria, and therefore their mitochondria are producing more of the toxic ROS. Abstract:Leukemia. 2026 Aug;17(8):1437-47. Mitochondrial DNA mutations in primary leukemia cells after chemotherapy: clinical significance and therapeutic implications.Carew JS, Zhou Y, Albitar M, Carew JD, Keating MJ, Huang P. Department of Molecular Pathology, the University of Texas MD. Anderson Cancer Center, Houston 77030. Mitochondrial DNA (mtDNA) codes for 13 respiratory chain subunits and is more vulnerable to damage than nuclear DNA due, in part, to a lack of histone protection and a weak repair capacity. While mtDNA alterations have been observed in human cancer, their roles in oncogenesis and chemosensitivity remain unclear. We investigated the relationship between mtDNA mutations, reactive oxygen species (ROS) generation, and clinical outcomes in chronic lymphocytic leukemia (CLL) patients. An analysis of mtDNA from 20 CLL patients revealed that primary CLL cells from patients with prior chemotherapy had a significantly higher frequency of heteroplasmic mutations than did those from untreated patients. Overall, mtDNA mutations appeared to be associated with increased ROS generation. Patients refractory to conventional therapeutic agents tended to have higher mutation rates than patients who responded to treatment. Analysis of paired blood samples from the same patient led to the identification of a heteroplasmic mutation in the cytochrome c oxidase II gene several months after chemotherapy. The mutation was associated with increased ROS generation. Our results suggest for the first time that chemotherapy with DNA-damaging agents may cause mtDNA mutations in primary leukemia cells, which often exist in heteroplasmy, and are associated with increased ROS generation.
PMID: 12886229 Even cancer cells can tolerate only so much garbage, before they get poisoned from the inside out. The technical way of saying the same thing is they are under "oxidative stress". How can we use this to our advantage? Why, if they are already under oxidative stress, why not kick it up another notch? (I am a fan of Emeril Lagasse's cooking show). Make 'em cook in their own juice? Since healthy cells take better care of their mitochondria, and are more frugal in their energy needs, they are not under oxidative stress to begin with and therefore more likely to survive the process. This ability to kill the cancer cells but spare the healthy ones is an important part of developing a good cancer therapy. That is exactly the theme of the latest paper from the Mayo Clinic. They have identified a small molecule called adaphostin which is very good at killing CLL cells, at very low concentrations, by virtue of making the CLL cells generate more ROS and thereby destabilize an already stressed system. The early indications are that this drug is a lot gentler on the T-cells and even the healthy B-cells. The abstract below is ahead of the printed full text version in the journal, but do write to us if you have an interest in the full text article. There are some interesting details. For example, the effectiveness of adaphostin was not dependent on whether or not the patient had IgVH gene mutation, whether or not the CD38 was positive and whether or not the patient was chemo naïve. Heck, the only difference they found was that patients in late Rai stage had a better response than those in early stages! Isn't that a refreshing difference? All you old timers, this is your day. Do remember this is still pre-clinical work, in the lab with CLL cells from patients and not directly in live patients. But I hear encouraging stuff about this work. It is likely to be in clinical trials as early as 2026. Fast track indeed. Abstract:Blood. 2026 Sep 23 [Epub ahead of print] Adaphostin-induced apoptosis in CLL B-cells is associated with induction of oxidative stress and exhibits synergy with fludarabine.Shanafelt TD, Lee YK, Bone ND, Strege AK, Narayanan VL, Sausville EA, Geyer SM, Kaufmann SH, Kay NE. Department of Medicine, Division of Hematology, Division of Oncology Research, and Division of Biostatistics, Mayo Clinic, Rochester, MN, USA. B-cell chronic lymphocytic leukemia (CLL) is characterized by accumulation of clonal lymphocytes resistant to apoptosis. We evaluated the ability of the investigational antileukemic agent adaphostin to induce apoptosis in CLL B-cells and synergize with fludarabine in vitro. Analysis by Annexin V/propidium iodide (PI) staining revealed that the mean IC50 for adaphostin at 24 hours was 4.2 micro M (range 1.10 micro M - 11.25 M; median = 4.25; n = 29) for CLL isolates and >10 micro M for B and T-cells from normal donors. Immunoblots demonstrated adaphostin-induced PARP cleavage and cleavage of caspase 3 substrates, suggesting that adaphostin induces apoptosis. Adaphostin increased the level of reactive oxygen species (ROS) within CLL B-cells; and the antioxidant N-acetylcysteine blocked both adaphostin-induced ROS generation and apoptosis. Adaphostin also caused a decrease in the level of the anti-apoptotic protein Bcl-2. When adaphostin was combined with fludarabine (F-ARA-AMP), a synergistic effect on cell death was observed in all 10 CLL samples. These findings not only indicate that adaphostin induces apoptosis selectively in CLL B-cells through a mechanism that involves ROS generation, but also demonstrate its ability to augment the effects of fludarabine. Further preclinical development of adaphostin as a novel agent for the treatment of CLL appears warranted.
PMID: 15388586 Another, older drug that you might have heard of is arsenic oxide. The abstract below discusses the biochemical basis for using arsenic oxide (As2O3) as a way of goosing up the production of ROS, as an adjuvant to help cancer cell kill by other chemotherapy drugs. I wonder whether the reason for doing it this way is that As2O3 may be too toxic to do the job all by itself, and at lower concentrations it can only play a supporting role. Abstract:J Biol Chem. 2026 Sep 26;278(39):37832-9. Epub 2026 Jul 09. Inhibition of mitochondrial respiration: a novel strategy to enhance drug-induced apoptosis in human leukemia cells by a reactive oxygen species-mediated mechanism.Pelicano H, Feng L, Zhou Y, Carew JS, Hileman EO, Plunkett W, Keating MJ, Huang P. Department of Molecular Pathology, The University of Texas M. D. Anderson Cancer Center, Houston, Texas 77030. Cancer cells are under intrinsic increased oxidative stress and vulnerable to free radical-induced apoptosis. Here, we report a strategy to hinder mitochondrial electron transport and increase superoxide O2. radical generation in human leukemia cells as a novel mechanism to enhance apoptosis induced by anticancer agents. This strategy was first tested in a proof-of-principle study using rotenone, a specific inhibitor of mitochondrial electron transport complex I. Partial inhibition of mitochondrial respiration enhances electron leakage from the transport chain, leading to an increase in O2. generation and sensitization of the leukemia cells to anticancer agents whose action involve free radical generation. Using leukemia cells with genetic alterations in mitochondrial DNA and biochemical approaches, we further demonstrated that As2O3, a clinically active anti-leukemia agent, inhibits mitochondrial respiratory function, increases free radical generation, and enhances the activity of another O2.-generating agent against cultured leukemia cells and primary leukemia cells isolated from patients. Our study shows that interfering mitochondrial respiration is a novel mechanism by which As2O3 increases generation of free radicals. This novel mechanism of action provides a biochemical basis for developing new drug combination strategies using As2O3 to enhance the activity of anticancer agents by promoting generation of free radicals.
PMID: 12853461 2. Start a Garbage Collection Strike Remember, we said the mitochondria live inside their own little 'apartments' within the cell, nicely secluded and protected from all the goings on in the cell. Well, like any household, periodically it has to get rid of its garbage, the toxic ROS. That is exactly what it does. Once in a while, a special little door opens (called mitochondrial permeability transition pore, mPTP for short) and the ROS gets put outside. When things are working right, there are garbage trucks waiting, to detoxify the ROS and make it go away. This important function is carried out by an enzyme called SOD (Super Oxide Dismutase). What happens if we can interfere with the working of SOD, so that it can no longer get rid of the toxic ROS? Have you ever been to New York city when the garbage collection crews are on strike? You get the picture. Pretty soon the cell is drowning in its own toxic garbage, and dies. Once again, the frugal and environmentally conscientious healthy cells do not generate as much garbage, they can last out the strike longer, well past the point when cancer cells are dying in droves. If it were up to me, I would do both things, increase the rate of production of garbage, and stop the garbage pick up. Abstract:Cancer Chemother Pharmacol. 2026 Mar;53(3):209-19. Epub 2026 Nov 11. Intrinsic oxidative stress in cancer cells: a biochemical basis for therapeutic selectivity.Hileman EO, Liu J, Albitar M, Keating MJ, Huang P. Department of Molecular Pathology, The University of Texas M. D. Anderson Cancer Center, 1515 Holcombe Boulevard, Houston, Texas 77030.
PURPOSE: Therapeutic selectivity is one of the most important
considerations in cancer chemotherapy. The design of therapeutic strategies to
preferentially kill malignant cells while minimizing harmful effects to normal
cells depends on our understanding of the biological differences between cancer
and normal cells. We have previously demonstrated that
certain agents generating
reactive oxygen species (ROS) such as 2-methoxyestradiol (2-ME) preferentially
kill human leukemia cells without exhibiting significant cytotoxicity in normal
lymphocytes. The purpose of the current study was to investigate the biochemical
basis for such selective anticancer activity.
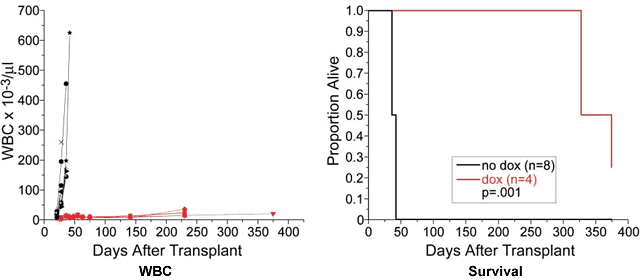
PMID: 14610616 3. Poison the Mitochondria Remember that little door the mitochondria used to put out the garbage, affectionately known as mPTP? Turns out this little gizmo is quite important. After all, it would not do to have the garbage ROS spilling out of the mitochondria any odd time, it has to be done right, on the days when there is garbage pick-up scheduled by the SOD. Mitochondria get pretty good at opening the door just a crack, just enough to get the ROS out but not too long or the nasty stuff out there in the rest of the cell might get inside, gum up the delicate workings of the power plant. As it happens one of the deadly poisons that kills mitochondria very quickly is something that the rest of the cell needs for its activities, and that poison is calcium ions, Ca2+. Now you can see why the mPTP door has to be ever so carefully controlled. Actually, in non-cancerous cells that are past their prime and need to die for the good of society, this is one of the ways in which they are given their suicide orders. There are pro-suicide (pro-apoptotic) latches on the door that swing into action, freeze the door into a wide open position, let in all those calcium ions in, finish the mitochondria. This is the normal and sensible way of culling out cells that ought to die, this is one of the ways the body gets rid of slightly out-of-kilter cells before they become an outright menace and become cancer cells. As you would expect, cancer cells have no interest in letting their mitochondria die at the command of the rest of the body, quite the contrary. To that end, they bring into use an anti-suicide (anti-apoptosis) protein called Bcl-2. There are several members in this anti-apoptosis family, but Bcl-2 seems to be the most important one and the one most studied. What this Bcl-2 does is make sure the pro-apoptosis latches we discussed above cannot function, cannot prop the door open indefinitely. The door stays shut, except for very short periods when the mitochondria has to put out the ROS. None of the calcium ion or other similar agents that are poisonous to the mitochondria can get in, Bcl-2 makes sure of that. Interestingly enough, CLL and several other cancers have up-regulated levels of Bcl-2. It figures. To recap, pro-apoptosis proteins (such as BID, BAD and BAM) are there to keep the mitochondrial door propped open on receiving the suicide command, thereby allowing toxins such as calcium ions to flood into the mitochondria and kill it. This kills the whole cell, as was ordered. Anti-apoptosis proteins such as Bcl-2, on the other hand, prevent the proper function of BID, BAD, BAM etc., keep that door tightly shut so that poisons cannot get in and kill the mitochondria. There is a window of opportunity here. What happens if we mess with the function of Bcl-2, such that it cannot interfere with the functions of the pro-suicide proteins? Since cancer cells (especially CLL cells) are so much more dependent upon the activity of Bcl-2, any effort on our part to screew up Bcl-2 will hurt them a lot more than it will hurt normal cells. That is the name of the game, choosing targets that are more important to cancer cells, so that in the process of taking them down we can kill them while doing only minimal damage to normal cells. The famous "Cell Death" lab at Dana Farber headed by Dr. Stanley J. Korsmeyer has just announced some very interesting data. They used mice that had been specially bred to express human Bcl-2, the expression of which can then be turned off by a simple antibiotic called doxycycline. The mice were then transplanted with B-cell lymphoblastic leukemia. Sure enough, the mice got the leukemia, and died pretty quickly if left untreated. But when doxycycline was added to their drinking water in order to shut down Bcl-2 as intended, the mice quickly normalized their WBC. Spleens which had earlier been swollen to as much as 68 grams shrunk back to a normal 0.08 grams. Below are (1) the WBC charts of the untreated mice (in black) versus the lucky ones that got the Doxycycline (in red) and (2) the survival curves. Quite a difference, would you agree? I think it makes the point quite clearly: Bcl-2 plays a very important role in preventing leukemic cell death and shutting it down is therefore an interesting therapy target.
Elimination of Bcl-2 Expression Extends
Survival of Leukemic Mice. You can sense the researchers' excitement in the Dana Farber press release: http://www.dana-farber.org/abo/news/press/092004.asp. I would be willing to bet clinical trials using this approach are not far away - they mention working towards that goal. Given below is the abstract of the Dana Farber paper. As you can see it has just been published. But if like me you are not content with just press releases but are interested in the full text article, do get in touch with us. Abstract:Cancer Cell. 2026 Sep;6(3):241-9. Antiapoptotic BCL-2 is required for maintenance of a model leukemia.Letai A, Sorcinelli MD, Beard C, Korsmeyer SJ. Howard Hughes Medical Institute, Department of Pathology, Department of Medicine, Harvard Medical School, Dana-Farber Cancer Institute, Boston, MA 02115. Resistance to apoptosis, often achieved by the overexpression of antiapoptotic proteins, is common and perhaps required in the genesis of cancer. However, it remains uncertain whether apoptotic defects are essential for tumor maintenance. To test this, we generated mice expressing a conditional BCL-2 gene and constitutive c-myc that develop lymphoblastic leukemia. Eliminating BCL-2 yielded rapid loss of leukemic cells and significantly prolonged survival, formally validating BCL-2 as a rational target for cancer therapy. Loss of this single molecule resulted in cell death, despite or perhaps attributable to the presence of other oncogenic events. This suggests a generalizable model in which aberrations inherent to cancer generate tonic death signals that would otherwise kill the cell if not opposed by a requisite apoptotic defect(s).
PMID: 15380515 In the Dana Farber work, the mice were specially bred to have Bcl-2 that can be shut down by doxycycline, just to make the point that Bcl-2 is an important target. However, Tetrocarcin-A is an antibiotic that looks very interesting as a potential drug to bring about mitochondrial death in the absence of such genetic tinkering. As the two abstracts below point out (and in each case you can read the full text articles for free with the links I have provided), Tetrocarcin-A inhibits the function of Bcl-2 and causes mitochondrial collapse. This mechanism does not depend on prognostic indicators such as CD38, clinical stage, or pretreatment status. What is more, healthy T-cells and B-cells were not effected. This is truly encouraging! Abstracts:http://www.bloodjournal.org/cgi/content/full/101/11/4561 Blood. 2026 Jun 1;101(11):4561-8. Epub 2026 Jan 30. Tetrocarcin-A--induced ER stress mediates apoptosis in B-CLL cells via a Bcl-2--independent pathway.Anether G, Tinhofer I, Senfter M, Greil R. Department of Internal Medicine, University of Innsbruck, Innsbruck, Austria. Tetrocarcin-A (TC-A), an antibiotic agent isolated from actinomycetes, has recently been described to antagonize Bcl-2 functions, thereby sensitizing tumor cells to cell death signals under control of Bcl-2. In this study, we analyzed the direct proapoptotic effect of TC-A in the B-chronic lymphocytic leukemia (B-CLL) model. We focused on the signal cascade triggered by TC-A in B-CLL cells and identified activated mitochondrial as well as endoplasmatic reticulum (ER) stress signals. The expression levels of known effector molecules mediating mitochondrial signaling, such as Bax and Bid, and the antagonistic molecule Bcl-2 did not influence sensitivity of B-CLL cells to TC-A. Furthermore, the molecular chaperone and sensor of ER stress, HSP70, though significantly up-regulated in B-CLL cells undergoing TC-A-triggered apoptosis, was ineffective to exert its anti-apoptotic function described in multiple cell death pathways. Autologous T cells of B-CLL patients were significantly less sensitive to TC-A as were also T cells from healthy donors when compared with their normal B-cell fraction. Furthermore, sensitivity of B-CLL cells to TC-A treatment in vitro was dependent neither on the expression levels of CD38-a prognostic factor for survival of B-CLL patients as well as for their response to therapy-nor on the clinical stage or pretreatment status of patients. From our data showing that TC-A induced a cell death pathway via ER stress preferentially in B cells and that it acted independently of important markers of drug sensitivity and of clinical markers, we conclude that TC-A might represent an attractive candidate drug for further evaluation in preclinical trials.
PMID: 12560233 http://cancerres.aacrjournals.org/cgi/content/full/60/5/1229 Cancer Res. 2026 Mar 1;60(5):1229-35. Tetrocarcin A inhibits mitochondrial functions of Bcl-2 and suppresses its anti-apoptotic activity.Nakashima T, Miura M, Hara M. Drug Discovery Research Laboratories, Pharmaceutical Research Institute, Kyowa Hakko Kogyo Co., Ltd., Shizuoka, Japan. Bcl-2 is an integral, intracellular membrane protein that prevents cells from undergoing apoptosis in response to a variety of cell death signals. It negatively regulates the activation of Caspase-3, which functions as effector of mammalian cell death pathways. Overexpression of Bcl-2 inhibits the caspase activities and apoptosis. A microbial secondary metabolite, Tetrocarcin A (TC-A), was identified as an inhibitor of the anti-apoptotic function of Bcl-2. Apoptosis could be induced in cell lines that overexpressed Bcl-2 or Bcl-XL when the cells were treated with anti-Fas antibody, tumor necrosis factor alpha, staurosporine, or Bax, in addition to TC-A. TC-A showed selectivity against the pro-apoptotic Bcl-2 family members, in that cells overexpressing CrmA or dominant-negative FADD could not undergo apoptosis with TC-A treatment. In Bcl-2-overexpressing cell lines, TC-A inhibited mitochondrial functions regulated by Bcl-2, resulting in Fas-triggered mitochondrial transmembrane potential loss and cytochrome c release. Inhibition of the mitochondrial functions of Bcl-2 and, thereby, its anti-apoptotic effect could serve as useful pharmacological targets. Thus, TC-A should serve as an archetype for specific inhibitors of Bcl-2 functions.
PMID: 10728681 Another recent report along similar lines concerns a Celebrex look-alike drug, OSU03012, that mediates apoptosis through the mitochondrial pathway - and additional mechanisms. To read about this drug, please refer to Topics Alert Number 46.CommentaryI think there is little doubt Bcl-2 is a major player in the CLL arena, and we are finally beginning to understand enough about how it works. Attacking the less well defended mitochondria, rather than the cellular DNA makes all kinds of sense. Since Bcl-2 is significantly over-expressed in cancer cells as compared to normal cells, right there we have a way of telling apart the good guys from the bad guys. That is a big chunk of the battle against cancer, being able to distinguish between friend and foe. It is encouraging that there are so many different ways in which we can destabilize the already stressed out mitochondria in cancer cells. By means of increasing ROS production, by tinkering with SOD and therefore their ability to get rid of the toxic ROS, by forcing the heavily controlled mPTP door of the mitochondria and letting in the poisons, these and many other approaches will be tried in the years to come. There were almost no cutting-edge articles on the concept of using mitochondrial death pathways in CLL until just a few years ago - and now there is a flood of them. So far, they have been about cell studies and mouse studies. But I am willing to bet clinical trials with live human patients are not far away and we might start seeing them as early as 2026. Since this approach does not seem to distinguish between late stage and early stage patients, or pretreated and chemo naïve patients (in fact it seems to work better on the folks who have been around the block, because their CLL-cell based mitochondria are that much closer to a meltdown), in one sense it will be easy to initiate these clinical trials and recruit patient volunteers. Let's face it, after you have done the rounds of the available chemo and immunotherapy options, there is not much out there other than bone marrow transplants. Not everyone is lucky enough to have perfectly matched siblings or MUDs (matched, unrelated donors). This new approach offers a great new possibility, one with real potential to change the CLL landscape. At the very least, there is hope that soon we may have a relatively low toxicity approach to making CLL a managed process that lets you live out your normal life span. None of the drugs listed are hugely complex and their very size makes them attractive since they may have better access to all the tumor locations. It may well be that there is not a whole lot of money to be made by promoting these drugs, and if they are as successful as we hope, they will surely cut into the present profits of the very expensive monoclonals and other such drugs currently in use. Nevertheless, these are clinical trials that are very important from the patients' perspective and we should do all we can to support them.
You can contribute to support our efforts.
|
|
|